Review Article
Volume 6 Issue 1 - 2024
A Comprehensive Review of the Pathological Mechanism of Evans Syndrome
Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research. Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, U.S.A
*Corresponding Author: Author (Escientific Publishers)*, Medical Genetics Director of the Division of Medical Genetics and Molecular Pathology Research. Division of Medical Genetics and Molecular Pathology Research, Center of Complex Disease, U.S.A
Received: April 03, 2024; Published: April 15, 2024
Abstract
Evans syndrome is a rare disease in which the immune system produces antibodies that mistakenly destroy red blood cells, platelets, and sometimes some white blood cells called neutrophils. This leads to abnormally low levels of these blood cells in the body (cytopenia). The premature destruction of red blood cells (hemolysis) is known as autoimmune hemolytic anemia, or AIHA. The symptoms and severity of Evans syndrome can be very different from person to person, as can the onset, course and duration of this disorder. Most people show a chronic course with periods of worsening of symptoms (exacerbation) and usually transient improvement with treatment. Most symptoms are caused by low levels of certain blood cells in the body. The exact cause of Evans syndrome is unknown. Evans syndrome is an autoimmune disease. It occurs when the immune system produces antibodies that mistakenly attack healthy tissue, particularly red blood cells, platelets, and sometimes some white blood cells.
Keywords: Evans Syndrome; Immune System Disorder; Hemolysis; Red Blood Cell
Overview of Evans Syndrome
Evans syndrome is a rare disease in which the immune system produces antibodies that mistakenly destroy red blood cells, platelets, and sometimes some white blood cells called neutrophils. This leads to abnormally low levels of these blood cells in the body (cytopenia). The premature destruction of red blood cells (hemolysis) is known as autoimmune hemolytic anemia, or AIHA. Thrombocytopenia refers to low levels of platelets (idiopathic thrombocytopenic purpura or ITP in this example). Neutropenia refers to low levels of certain white blood cells known as neutrophils. Evans syndrome is defined as the association between AIHA and ITP. Neutropenia occurs less often. In some cases, autoimmune destruction of these blood cells occurs simultaneously. In most cases, one disease develops first before another condition develops later (sequentially). The symptoms and severity of Evans syndrome can vary greatly from person to person. Evans syndrome can potentially cause serious and life-threatening complications. Evans syndrome can occur alone as a primary disorder (idiopathic) or in association with other autoimmune disorders or lymphoproliferative disorders as a secondary disorder. (Lymphoproliferative disorders are characterized by an overproduction of white blood cells.) The distinction between primary and secondary Evans syndrome is important because it can affect treatment.[1]
Evans syndrome is a rare disease in which the immune system produces antibodies that mistakenly destroy red blood cells, platelets, and sometimes some white blood cells called neutrophils. This leads to abnormally low levels of these blood cells in the body (cytopenia). The premature destruction of red blood cells (hemolysis) is known as autoimmune hemolytic anemia, or AIHA. Thrombocytopenia refers to low levels of platelets (idiopathic thrombocytopenic purpura or ITP in this example). Neutropenia refers to low levels of certain white blood cells known as neutrophils. Evans syndrome is defined as the association between AIHA and ITP. Neutropenia occurs less often. In some cases, autoimmune destruction of these blood cells occurs simultaneously. In most cases, one disease develops first before another condition develops later (sequentially). The symptoms and severity of Evans syndrome can vary greatly from person to person. Evans syndrome can potentially cause serious and life-threatening complications. Evans syndrome can occur alone as a primary disorder (idiopathic) or in association with other autoimmune disorders or lymphoproliferative disorders as a secondary disorder. (Lymphoproliferative disorders are characterized by an overproduction of white blood cells.) The distinction between primary and secondary Evans syndrome is important because it can affect treatment.[1]
Evans syndrome was first described in the medical literature in 1951 by Dr. Robert Evans and colleagues. For many years this disorder was considered a random event of AIHA with thrombocytopenia or neutropenia. However, researchers now believe that this disorder represents a distinct condition characterized by chronic and profound dysfunction (dysregulation) of the immune system (more so than ITP or AIHA alone). [1]
Clinical Signs and Symptoms of Evans Syndrome
The symptoms and severity of Evans syndrome can be very different from person to person, as can the onset, course and duration of this disorder. Most people show a chronic course with periods of worsening of symptoms (exacerbation) and usually transient improvement with treatment. Most symptoms are caused by low levels of certain blood cells in the body. These blood cells perform specific tasks. Red blood cells supply the body with oxygen and remove carbon dioxide, platelets help clot to stop blood loss, and white blood cells help fight infections. [1]
The symptoms and severity of Evans syndrome can be very different from person to person, as can the onset, course and duration of this disorder. Most people show a chronic course with periods of worsening of symptoms (exacerbation) and usually transient improvement with treatment. Most symptoms are caused by low levels of certain blood cells in the body. These blood cells perform specific tasks. Red blood cells supply the body with oxygen and remove carbon dioxide, platelets help clot to stop blood loss, and white blood cells help fight infections. [1]

Figure 1: Schematic of the physiological mechanism of the immune system in Evans syndrome. [1]
Some people with Evans syndrome may initially destroy red blood cells faster than the body can replace them. Low levels of circulating red blood cells, known as anemia, can cause a variety of symptoms, including fatigue, pale skin (pallor), dizziness, shortness of breath, dark urine, and palpitations. Some people may experience yellowing of the skin and especially the whites of the eyes (jaundice). [1,2]
Other people may initially present with low platelet levels, known as thrombocytopenia. Thrombocytopenia can cause tiny reddish or purple spots on the skin (petechiae), a broader purplish discoloration of the skin caused by bleeding from broken blood vessels into the tissue under the skin (ecchymosis), and purpura, a skin rash made up of spots. internal bleeding from the small area of blood vessels. Affected individuals may be more prone to bruising following minor injuries and spontaneous bleeding from the mucous membranes. [1,2]
Low levels of white blood cells, known as neutropenia, are less common in people with Evans syndrome than anemia or thrombocytopenia. People with neutropenia may be prone to frequent infections. General symptoms may include fever, general malaise (malaise), and sores on the mucous membranes of the mouth. [1,2]
Additional symptoms that may occur in people with Evans syndrome include swollen lymph nodes, spleen, and liver. These results may come and go or in some cases may only occur in acute periods. In most cases, patients with Evans syndrome may not respond to treatment (resistant Evans syndrome) and can eventually lead to life-threatening complications, including sepsis, severe bleeding (bleeding) episodes, and reversible cardiovascular problems, including l heart failure. [1,2]
Etiology of Evans Syndrome
The exact cause of Evans syndrome is unknown. Evans syndrome is an autoimmune disease. It occurs when the immune system produces antibodies that mistakenly attack healthy tissue, particularly red blood cells, platelets, and sometimes some white blood cells. [1,3]
The exact cause of Evans syndrome is unknown. Evans syndrome is an autoimmune disease. It occurs when the immune system produces antibodies that mistakenly attack healthy tissue, particularly red blood cells, platelets, and sometimes some white blood cells. [1,3]
The immune system usually responds to foreign substances by producing specialized proteins called antibodies. Antibodies work by directly destroying foreign substances or by coating them with substances that mark them for destruction by white blood cells. When antibodies target healthy tissue, they may be called autoantibodies. Researchers believe that a trigger event (such as an infection or underlying disorder) may cause the immune system to produce autoantibodies in Evans syndrome. [1,3]
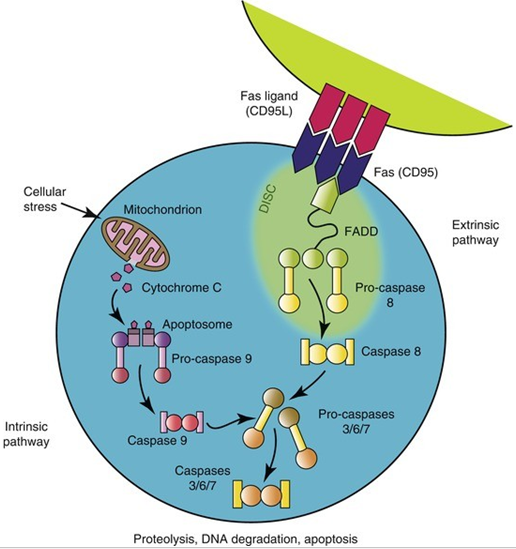
Figure 2: Schematic of the pathway, biochemical mechanism, and pathogenesis of Evans syndrome. [1]
Evans syndrome can occur with another disorder as a secondary disease. Secondary Evans syndrome may be associated with other disorders such as autoimmune lymphoproliferative syndrome (ALPS), lupus, antiphospholipid syndrome, Sjogren's syndrome, common variable immunodeficiency, IgA deficiency, specific lymphoma, and chronic lymphocytic leukemia. [1,3]
Frequency of Evans Syndrome
The incidence and prevalence of Evans syndrome are unknown. This disorder can affect children or adults. [1,4]
The incidence and prevalence of Evans syndrome are unknown. This disorder can affect children or adults. [1,4]
Disorders Associated with Evans Syndrome
Symptoms of the following disorders may be similar to the symptoms of Evans syndrome. The comparison can be useful for differential diagnosis :
Symptoms of the following disorders may be similar to the symptoms of Evans syndrome. The comparison can be useful for differential diagnosis :
Several disorders can be characterized by the presence of hemolytic anemia and thrombocytopenia. These disorders include paroxysmal nocturnal hemoglobinuria (PNH), acquired thrombotic thrombocytopenic purpura, hemolytic uremic syndrome, and Kasabach-Merritt syndrome. [1,4]
Autoimmune lymphoproliferative syndrome (ALPS) is a rare genetic disorder that overlaps with Evans syndrome. A characteristic finding of ALPS is the presence of abnormally high numbers of white blood cells called lymphocytes, which can accumulate in the lymph nodes, liver, and spleen and cause enlargement of these organs. ALPS can lead to symptoms similar to Evans syndrome, particularly anemia, thrombocytopenia, and neutropenia. Most people with ALPS have a mutation in the tumor necrosis factor receptor superfamily member gene (TNFRSF6), also called CD95 or Fas. The exact relationship, if any, between these disorders is not fully understood. It is believed that approximately half of children diagnosed with Evans syndrome may have an underlying ALPS cause because autoreactive cells do not undergo normal programmed cell death (apoptosis). The intersection of ALPS and Evans syndrome in adults is less clear. [1,4]
Diagnosis of Evans Syndrome
The diagnosis of Evans syndrome is based on the identification of characteristic symptoms, a detailed patient history, a comprehensive clinical evaluation and a series of specialized tests. There is no specific test for Evans syndrome and it is diagnosed after other possible diagnoses have been ruled out. Specifically, a diagnosis of Evans syndrome can be made when autoimmune hemolytic anemia (with positive direct Coombs test) and thrombocytopenia (ITP) occur in the same patient, even if they are not concurrent. [1,4]
The diagnosis of Evans syndrome is based on the identification of characteristic symptoms, a detailed patient history, a comprehensive clinical evaluation and a series of specialized tests. There is no specific test for Evans syndrome and it is diagnosed after other possible diagnoses have been ruled out. Specifically, a diagnosis of Evans syndrome can be made when autoimmune hemolytic anemia (with positive direct Coombs test) and thrombocytopenia (ITP) occur in the same patient, even if they are not concurrent. [1,4]
Laboratory studies include a complete blood count (CBC), which may show low levels of red blood cells, platelets, and white blood cells. Another blood test, known as a direct antiglobulin test, or DAT, is used to determine whether the amount of certain antibodies is higher than normal. A series of tests can be performed to rule out other conditions. Such tests may include bone marrow biopsy, additional antibody tests, and CT scans of the chest, abdomen, and pelvis. [1,4]
Some doctors recommend that children with Evans syndrome be screened for ALPS because the prevalence of these two disorders is high. Screening includes testing for the presence of double negative T cells (DNT) by flow cytometry, the presence of which indicates ALPS. [1,5]
Treatment Paths for Evans Syndrome
There is no cure for Evans syndrome, and treatment is often challenging. Treatment is directed at specific symptoms evident in each individual. Treatment may require the coordinated effort of a team of specialists. Pediatricians, surgeons, hematologists, pediatric hematologists, immunologists, rheumatologists and other healthcare professionals may need to systematically and comprehensively plan an effective treatment for a child. [1,5]
There is no cure for Evans syndrome, and treatment is often challenging. Treatment is directed at specific symptoms evident in each individual. Treatment may require the coordinated effort of a team of specialists. Pediatricians, surgeons, hematologists, pediatric hematologists, immunologists, rheumatologists and other healthcare professionals may need to systematically and comprehensively plan an effective treatment for a child. [1,5]
Most affected individuals require treatment, although spontaneous recovery has been reported in rare cases. Several different types of treatments have been used to treat people with Evans syndrome, and their effectiveness has varied greatly among affected people. Some people have long-term recovery from this disorder. Others experience chronic problems without improvement. [1,6]
As a result, treatment procedures and specific interventions may vary based on several factors, including the severity of the disease. blood cell count level; presence or absence of specific symptoms; the person's age and general health; and/or other elements Decisions regarding the use of specific drug regimens or other treatments should be made by physicians and other members of the healthcare team in careful consultation with the patient based on the characteristics of his or her case. [1,6]
First-line treatment for Evans syndrome often includes corticosteroids such as prednisolone. Corticosteroids help suppress the immune system and reduce the production of autoantibodies. The first results are often effective. Intravenous immunoglobulin (IVIg) therapy has also been used to treat people with Evans syndrome. IVIg treatment modifies the activity of the immune system. IVIg is a solution containing antibodies donated by healthy people and injected directly into the vein. [1,7]
Surgical removal of the spleen (splenectomy) has been used to treat some people with Evans syndrome. Most reports are non-definitive and anecdotal, and it is difficult to determine the effectiveness of this method due to the simultaneous use of other treatments such as medications. Splenectomy is generally reserved for people who do not respond to other treatment options (refractory Evans syndrome). In children, splenectomy rarely results in long-term improvement in symptoms. In adults, effectiveness varies and symptoms usually return at some point. Therefore, it is usually delayed as much as possible and every effort is made not to do so. [1,7]
During an acute episode, blood or platelet transfusions may be needed to relieve symptoms. However, the use of blood or platelet transfusions should be avoided as much as possible. [1,7]
New treatments for Evans syndrome are being studied. Rituximab appears to be a very effective treatment for patients with Evans syndrome. Rituximab is classified as a monoclonal antibody or biologic therapy, drugs that act like antibodies but are created artificially in a laboratory. Preliminary studies have shown this drug to be generally safe and effective. The advantages of rituximab are to avoid severe immunosuppression and side effects associated with other immunosuppressive agents. The disadvantage is that in cases of Evans syndrome caused by underlying ALPS, hypogammaglobulinemia appears to develop which may persist in patients treated with rituximab. Hypogammaglobulinemia is a condition in which the body's immune system does not produce enough antibodies and potentially makes those affected susceptible to bacterial infections and, to some extent, some viral infections. [1,7]
Additional drugs have been studied in a small number of people with Evans syndrome. The second factor that appears to be relatively effective in this disorder is mycophenolate mofetil. These drugs can be used alone or in combination (multi-agent therapy) as second-line treatments for people with Evans syndrome who do not respond to corticosteroids or IVIg therapy. More research is needed to determine the long-term safety and effectiveness of these potential treatments for people with Evans syndrome. [1,7]
Research treatments
Some people with Evans syndrome have been treated with allogeneic or autologous stem cell transplantation. Stem cells are special cells that are found in the bone marrow and produce different types of blood cells (ie red blood cells, white blood cells and platelets). In an autologous stem cell transplant, the patient's stem cells are removed after previous treatment, usually with drugs. These healthy stem cells are re-injected into the bone marrow after the progression of the disorder. In an allogeneic stem cell transplant, stem cells are donated from another person, usually a close family member. These methods are usually used as a last resort for people who have failed to respond to other forms of treatment. They have produced mixed results, and more experience is necessary to determine their potential effectiveness as a treatment for Evans syndrome. [1,7]
Some people with Evans syndrome have been treated with allogeneic or autologous stem cell transplantation. Stem cells are special cells that are found in the bone marrow and produce different types of blood cells (ie red blood cells, white blood cells and platelets). In an autologous stem cell transplant, the patient's stem cells are removed after previous treatment, usually with drugs. These healthy stem cells are re-injected into the bone marrow after the progression of the disorder. In an allogeneic stem cell transplant, stem cells are donated from another person, usually a close family member. These methods are usually used as a last resort for people who have failed to respond to other forms of treatment. They have produced mixed results, and more experience is necessary to determine their potential effectiveness as a treatment for Evans syndrome. [1,7]
Discussion
Evans syndrome was first described in the medical literature in 1951 by Dr. Robert Evans and colleagues. For many years this disorder was considered a random event of AIHA with thrombocytopenia or neutropenia. However, researchers now believe that this disorder represents a distinct condition characterized by chronic and profound dysfunction (dysregulation) of the immune system (more so than ITP or AIHA alone). Additional symptoms that may occur in people with Evans syndrome include swollen lymph nodes, spleen, and liver. These results may come and go or in some cases may only occur in acute periods. In most cases, patients with Evans syndrome may not respond to treatment (resistant Evans syndrome) and can eventually lead to life-threatening complications, including sepsis, severe bleeding (bleeding) episodes, and reversible cardiovascular problems, including l heart failure. Some doctors recommend that children with Evans syndrome be screened for ALPS because the prevalence of these two disorders is high. Screening includes testing for the presence of double negative T cells (DNT) by flow cytometry, the presence of which indicates ALPS. New treatments for Evans syndrome are being studied. Rituximab appears to be a very effective treatment for patients with Evans syndrome. Rituximab is classified as a monoclonal antibody or biologic therapy, drugs that act like antibodies but are created artificially in a laboratory. Preliminary studies have shown this drug to be generally safe and effective. The advantages of rituximab are to avoid severe immunosuppression and side effects associated with other immunosuppressive agents. The disadvantage is that in cases of Evans syndrome caused by underlying ALPS, hypogammaglobulinemia appears to develop which may persist in patients treated with rituximab. Hypogammaglobulinemia is a condition in which the body's immune system does not produce enough antibodies and potentially makes those affected susceptible to bacterial infections and, to some extent, some viral infections z. [1-7]
References
- Asadi S, (2024). Human Idiopathic Diseases Books, Amidi Publications, Iran.
- Ware RE. (2009). Autoimmune Hemolytic Anemia. In: Nathan and Oski’s Hematology of Infancy and Childhood, 7th ed. Orkin SH, Nathan DG, Ginsburg D, Look AL, Fisher DE, Lux SE, editors. Saunders Elsevier, Philadelphia, PA. pp. 633-634.
- Carey EJ, Somaratne K, Rakela J. (2011). Successful rituximab therapy in refractory autoimmune hepatitis and Evans syndrome. Rev Med Chil. 139: 1484-1487.
- Farruggia P, Macaluso A, Tropia S, et al. (2011). Effectiveness of cyclosporine and mycophenolate mofetil in a child with refractory Evans syndrome. Pediatr Rep. 3: e15.
- Seif AE, Manno CS, Sheen C, Grupp SA, Teachey DT. (2010). Identifying autoimmune lymphproliferative syndrome in children with Evans syndrome: a multi-institutional study. Blood. 115: 2142-2145.
- Michel M, Chanet V, Dechartes A, et al. (2009). The spectrum of Evans syndrome in adults: new insight into the disease based on analysis of 68 cases. Blood. 114: 3167-3172.
- Mathew P. Evans Syndrome. Emedicine Journal, March 12, (2012). Available at: http://emedicine.medscape.com/article/955266-overview Accessed on: February 18, 2013.
Citation: Author (Escientific Publishers)*. (2024). A Comprehensive Review of the Pathological Mechanism of Evans Syndrome. Journal of Biotechnology and Immunology 6(1).
Copyright: © 2024 Author (Escientific Publishers)*. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.