Research Article
Volume 3 Issue 1 - 2021
Theoretical Study of the Reduction Reaction and Conformational Analysis of Paraquat by Density Functional Theory
Joacy Vicente Ferreira, Instituto Federal de Pernambuco, Campus Afogados da Ingazeira, Rua Edson Barbosa de Araújo, s/n, Bairro Manoela Valadares 56800-000, Afogados da Ingazeira - Pernambuco, Brasil
*Corresponding Author: Joacy Vicente Ferreira, Instituto Federal de Pernambuco, Campus Afogados da Ingazeira, Rua Edson Barbosa de Araújo, s/n, Bairro Manoela Valadares 56800-000, Afogados da Ingazeira - Pernambuco, Brasil.
Received: August 05, 2021; Published: August 16, 2021
Abstract
Calculations of the reduction reaction and conformations of the paraquat have been studied theoretically within the method of density functional theory using the B3LYP functional and 6-31++G(d,p) basis sets. Through of the conformational analysis it was possible to evaluate the relative stability of the conformational isomers obtained by rotating around the C4C3 bond, from systematic variations of the C17C4C3C2 dihedral. The paraquat is found to be twisted about the inter-ring bond. With results of lowest unoccupied molecular orbital and natural bond orbital atomic partial charges it was possible to identify the atoms N7 or N19 as is the most likely region for reduction and a mechanism has been proposed.
Keywords: DFT; Paraquat; Reduction; Conformational
Abbreviations: HOMO: highest occupied molecular orbital; LUMO: lowest unoccupied molecular orbital; DFT: density functional theory; HF: Hartree Fock; NBO: natural bond orbital; B3LYP: Becke, three-parameter; EA: electron affinity; ESCF: self-consistent field energy.
Introduction
The modern agriculture is heavily dependent on the use of pesticides as protective agents against all sorts of plant diseases. The paraquat (1, 1’-dimethyl-4,4’-bipyridylium ion) whose molecular structure is shown in Figure 1 is the most toxic herbicide, the third most widely used in the world, almost more than 120 countries and used for more than 50 plants for both terrestrial and aquatic [1, 2, 3].
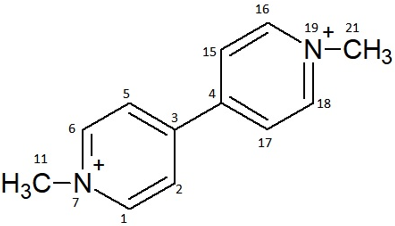
Figure 1: Structure of the paraquat.
Conformational analysis is the study of molecular conformations and their influences on the physicochemical properties of this molecule. This analysis is usually done by performing a rotation around a single bond, varying the dihedral angle of the bonds. The conformational preference of bipyridiniums depend on electronic structure [4] and according to Calderbrank [5] has been implicated as a property which determines their suitability as electron relays.
Through the atomic charges and frontier molecular orbitals: HOMO and LUMO [6], giving an estimate of likely atoms involved process of oxidation or reduction, and represent the ability of a chemical to donate or accept electrons. The HOMO energy is directly related to the ionization potential, while the LUMO energy is related to the electron affinity.
We conducted DFT conformational analyses and calculations of determining exactly the atoms where occurs the reduction the likely mechanism of paraquat. A theoretical study similar was used successfully on the proposition of the mechanism of reduction of berenil [7].
Computational Details
All Calculations on the paraquat molecule in the ground state and reduced form have been performed using DFT with the B3LYP functional [8, 9] and the 6-31+G(d,p) basis set as implemented in the Gaussian 09 program [10].
All Calculations on the paraquat molecule in the ground state and reduced form have been performed using DFT with the B3LYP functional [8, 9] and the 6-31+G(d,p) basis set as implemented in the Gaussian 09 program [10].
The electron affinity can be obtained by two procedures: the first through Koopmans′ theorem states that the EA [11] can be expressed in terms of the ELUMO (Eq. 1) and the second through ΔESCF method [12] (Eq. 2).
EA = –ELUMO, (1)
EA = ESCF (N electrons) – ESCF (N + 1 electrons), (2)
Results and Discussion
Geometric analysis
The optimized energy (total energy of the paraquat) calculations, in atomic units is -574.6903186. The most stable state that the total energy from HF and DFT calculations (-570.966455395 a.u and -569.609154759 a.u respectively) obtained of Kreisig [3].
The optimized energy (total energy of the paraquat) calculations, in atomic units is -574.6903186. The most stable state that the total energy from HF and DFT calculations (-570.966455395 a.u and -569.609154759 a.u respectively) obtained of Kreisig [3].
The geometric optimization is an important step for obtaining correct of the partial atomic charges, frontier molecular orbitals and conformers isomers. The Table 1 shows the molecular geometry parameter of the paraquat. The calculated bond length (in Å) and bond angles (in°) is in a good agreement with the experimental values [13].
| Parameter | Bond length | Experimental |
| N7-C11 | 1.49 | 1.47 |
| N7-C6 | 1.35 | 1.36 |
| N7-C1 | 1.35 | 1.34 |
| C6-C5 | 1.39 | 1.37 |
| C5-C3 | 1.40 | 1.38 |
| C3-C2 | 1.41 | 1.41 |
| C2-C1 | 1.38 | 1.38 |
| C3-C4 | 1.49 | 1.49 |
| C4-C15 | 1.40 | 1.38 |
| C15-C16 | 1.39 | 1.37 |
| N19-C16 | 1.35 | 1.36 |
| N19-C18 | 1.35 | 1.34 |
| N19-C21 | 1.49 | 1.47 |
| C18-C17 | 1.38 | 1.38 |
| C17-C4 | 1.41 | 1.41 |
| Parameter | Bond angle | Experimental |
| N7-C6-C5 | 121 | 119-121 |
| C6-C5-C3 | 120 | 119-122 |
| C5-C3-C2 | 118 | 117-120 |
| C3-C2-C1 | 120 | 119-122 |
| C2-C1-N7 | 121 | 119-121 |
| C6-N7-C1 | 120 | 119-123 |
| N19-C16-C15 | 121 | 119-121 |
| C16-C15-C4 | 120 | 119-122 |
| C15-C4-C17 | 118 | 117-120 |
| C4-C17-C18 | 120 | 119-122 |
| C17-C18-N19 | 121 | 119-121 |
| C18-N19-C16 | 120 | 119-123 |
Table 1: Geometrical parameters calculated for paraquat.
Conformational analysis
The rotational energy barrier related to the rotation around the C3C4 bond was investigated in paraquat. From systematic variations of the C17C4C3C2 dihedral angle, the relative energy curve (Figure 2) showed the existence of a minimum structure (more stable conformational isomer) and a maximum structure (less stable conformational isomer). The planar structure (0°) is estabilized by resonance interactions between the two aromatic rings and resides on top of maximum. The orthogonal structure (90°) also represents a maximum. This potential curve exhibiting a minimum at 41° very similar to 40° obtained by Kleier [4].
The rotational energy barrier related to the rotation around the C3C4 bond was investigated in paraquat. From systematic variations of the C17C4C3C2 dihedral angle, the relative energy curve (Figure 2) showed the existence of a minimum structure (more stable conformational isomer) and a maximum structure (less stable conformational isomer). The planar structure (0°) is estabilized by resonance interactions between the two aromatic rings and resides on top of maximum. The orthogonal structure (90°) also represents a maximum. This potential curve exhibiting a minimum at 41° very similar to 40° obtained by Kleier [4].
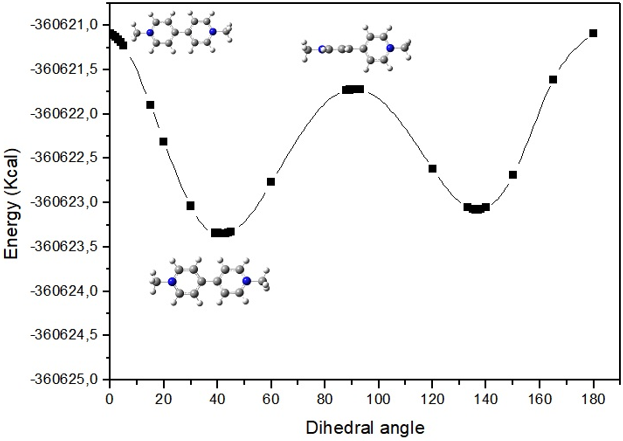
Figure 2: Torsional potential for paraquat.
Reduction Reaction
The reduction of paraquat showed a reaction with the entry 1e- [5]. An electron is added to the paraquat (charge +2) yielding reduced paraquat (charge +1).
The reduction of paraquat showed a reaction with the entry 1e- [5]. An electron is added to the paraquat (charge +2) yielding reduced paraquat (charge +1).
The EA was calculated by Koopmans′ theorem (-ELUMO = 236.96 Kcal/mol) and by the ΔESCF method (207.74 Kcal/mol) by difference between the total energy of the paraquat (charge +2) and the paraquat (charge +1). In this case, the paraquat (charge +2) was been optimized at the same level of theory as the paraquat (charge +1). These results show the stability of the reduction reaction and according to Sadi [14] the procedure second that was chose gives more accurate results that Koopmans approach.
Kleier [4] suggest that the reduction occurs (C3-C4 carbons) of the paraquat molecule. However, they don’t show the mechanism of the reduction reaction. In order to identify the most probable sites for the reduction reaction of the molecule, a LUMO contour map (Figure 3) analysis are obtained for paraquat in the ground state (charge +2).
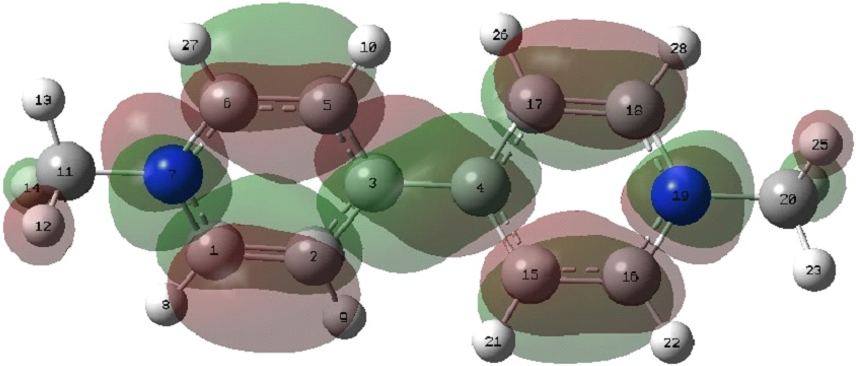
Figure 3: LUMO contour map of the paraquat in the ground state.
The orbital LUMO provides evidence on the sites of reduction of a molecule as this orbital is responsible for entry of electrons. As can be seen in the Figure 3, the orbitals located in the atoms C1, C2, C3, C4, C5, C6, N7, C15, C16, C17, C18 and N19 offer contributions to form the LUMO. Hence, those regions must be directly involved in the reduction process.
To realm the evidences observed from the LUMO analysis, NBO atomic charges [15] were calculated for two states paraquat: ground state (charge +2) and reduced form (charge +1). This calculation allowed evaluating the change in electron density before and after the reduction of the molecule. The values of the atomic charge calculated are indicated in Table 2.
| Atoms | Paraquat (+2) | Paraquat (+1) |
| C1 | 0.09 | 0.03 |
| C2 | -0.21 | -0.24 |
| C3 | 0.01 | -0.07 |
| C4 | 0.01 | -0.02 |
| C5 | -0.21 | -0.22 |
| C6 | 0.10 | 0.02 |
| N7 | -0.29 | -0.36 |
| C15 | -0.21 | -0.25 |
| C16 | 0.10 | 0.03 |
| C17 | -0.21 | -0.25 |
| C18 | 0.09 | 0.03 |
| N19 | -0.29 | -0.36 |
Table 2: Calculated atomic charges for the paraquat molecule in the ground state and reduced form.
The Table 2 shows that the inclusion of one electron in the paraquat (+2), leads to an increase in the electronic density of the atoms C1, C2, C3, C4, C5, C6, N7, C15, C16, C17, C18 and N19. As C3 and C6 atoms, are located in aromatic rings, the delocalization of charges in these regions explains the variations found out thus they are not expected to represent sites of reduction. It is also observed in Table 2 the largest variations in charge density occurred in the N7 or N19 (-0.29 in the ground state and -0.36 after reduction). Thus, in the mechanism is the entrance of an electron in the atom N7 or N19 in the sequence there is a rearrangement and the electron is then transferred to one of the pyridinium rings yelding a free radical (scheme 1).
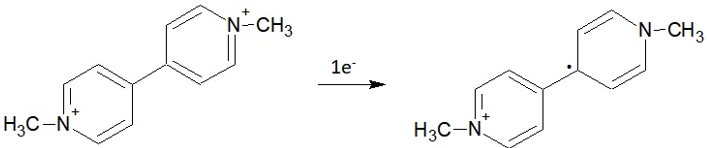
Scheme 1: Paraquat reduction mechanism.
Conclusion
The conformational analysis it was possible to evaluate the relative stability of the conformational isomers. These calculations conformations show preference for a twisted conformational isomer (41o) rather than a planar structure isomer.
The LUMO contour map of the paraquat and the atomic charges of the paraquat molecule in the ground state and after the reduction, reveals large variations in charge densities for nitrogen atoms, indicating these atoms as the site of reduction in the molecule. A possible mechanism for the reaction of reduction of paraquat was suggested.
Acknowledgments
The authors thank the research groups LQTC of the Federal University of Pernambuco and CENAPAD-SP.
The authors thank the research groups LQTC of the Federal University of Pernambuco and CENAPAD-SP.
References
- Arfi F, Safni, Abdullah Z, (2017). Degradation of paraquat in gramoxone pesticide with addition of ZnO. Molekul 12: 159-165.
- Suntres ZE, (2002). Role of antioxidants in paraquat toxicity. Toxicology 180: 65-77.
- Kreisig S, Tarzona A, Koglin E, (1997). Thead sorption of paraquat on silver electrode surfaces: a SERS microprobe study. Electrochemical acta 42: 3335-3344.
- Kleier DA, Weeks GH, (1986). Electronic structure and conformational analysis of paraquat in three oxidation states. Journal of Molecular Structure (Theochem) 148: 25-31.
- Calderbank A, Tahori S. (1972), Pestic. Chem., Proc. 2nd Int. Conf. Pestic. Chem., Vol. 5, Gordon and Breach, New York.
- Clare BW, (1994). Frontier orbital energies in quantitative structure-activity relationships: A comparison of quantum chemical methods. Theoretica Chimica Acta 87: 415-430.
- Ferreira JV, Abreu FC, Pavão AC, Nascimento VB, (2017). Theoretical Study and the Mechanism for the Berenil Reduction Reaction. Research & Reviews: Journal of Chemistry 6: 60-63.
- Lee C, Yang W, Parr RG, (1988). Development of the Colle-Salvetti correlation-energy formula into a functional of the eléctron density. Physical Review. B 37: 785.
- Jordá JMP, Becke AD, (1995). A density-functional study of van der Waals forces: rare gas diatomic. Chemical Physics Letters233: 134-137.
- Gaussian 09, Revision A.03. Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JA Jr, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Cliford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA (2003) Gaussian 09 (Revision A.7). Gaussian Inc., Pittsburgh.
- Koopmans T, (1934). Über Die Zuordnung Von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms. Physica 1: 104-113.
- Chattaraj PK, Duley S, (2010). Electron Affinity, Electronegativity, and Electrophilicity of Atoms and Ions, J. Chem. Eng. Data 55: 1882-1886.
- Rust PM, (1975). N,N’-Dimetryl-4,4’-bibyridylium (paraquat) hexachlorodicuprate (II). Acta Crystallographica section B 31: 1771-1772.
- Sadi A, Ouamerali O, (2020). Theoretical study, NBO analysis, HOMO/LUMO and First Static hyperpolarizability for the structural prediction of new 1X-tri-R-σ3 λ3 –Phosphacyclohexadienyl and anion ligands using DFT calculations. Journal of Structural Chemistry 61: 166-181.
- Red AE, Weinstock RB, Weinhold F, (1985). Natural population analysis.Journal of Chemical Physics 83: 735.
Citation: Joacy Vicente Ferreira. (2021). Theoretical Study of the Reduction Reaction and Conformational Analysis of Paraquat by Density Functional Theory. Archives of Chemistry and Chemical Engineering 3(1).
Copyright: © 2021 Joacy Vicente Ferreira. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.