Review Article
Volume 2 Issue 1 - 2020
New Evolutionary Scenario and Molecular Diagnostic Methods in typing of Mycobacterium Tuberculosis Complex strains
Ethiopian institute of Agricultural Research, Holeta Agricultural Research Center, Oromia Region, Ethiopia
*Corresponding Author: Beksisa Urge Hurrisa, Ethiopian institute of Agricultural Research, Holeta Agricultural Research Center, Oromia Region, Ethiopia.
Received: February 08, 2020; Published: May 04, 2020
Abstract
Tuberculosis is one of the major health problems responsible for morbidity and mortality particularly in developing countries. It is a bacterial disease caused by a group of closely related species of Mycobacterium tuberculosis complex such as M. tuberculosis, Mycobacterium Canetti, Mycobacterium Africanum, Mycobacterium microti, M. bovis, Mycobacterium caprae and Mycobacterium pinnipedii have genetic similarity. The Mycobacterium Tuberculosis Complex (MTBC) species are closely related taxonomic group of bacteria contributing to hundred-percent chromosomal homology between each other. The genome of MTBC is about 4.4 M bp and contains 0.01-0.3 percent synonymic nucleotide polymorphisms. It is characterized by 99.9% similarity at the nucleotide level and identical 16S rRNA sequences but differs widely in host related tropisms, phenotypes, and pathogenicity. MBTC strains have emerged from a common origin through DNA deletion and insertion mechanisms which resulted in the present Mycobacterium species. These insertion –deletion in the variable region of genomes or region of difference (RD) of the MBTC is used to identify the phylogenetic relationships between strains of the M. tuberculosis complex and M. tuberculosis. The deletion of genomic or DNA region reflected RD or evolutionary lineages of M. africanum, M. microti and M. bovis that diverged from the progenitor of the present M. tuberculosis strains. The presence of conservation in the house keeping genes of the species of M. tuberculosis complex causes a rapid evolutionary and emerging processes. There are advanced genotyping tools that are widely used to differentiate Mycobacterium tuberculosis strains. The widely used diagnostic methods are; IS6110 restriction fragment length polymorphism, Spoligotyping, and mycobacterial interspersed repeat units variable number of tandem repeats. These techniques would help for understanding molecular mechanisms and identification of mycobacterial strains to institute the nature of their diversity on the phylogenetic tree that could aid to know the evolutionary situation of emerging strains. Therefore, the need to adopt advanced molecular tools to decipher circulating strains of MTBC should be encouraged.
Keywords: Tuberculosis; Genetic diversity; Mycobacterium Tuberculosis complex; Region of difference
Introduction
Tuberculosis is a bacterial disease caused by Mycobacterium tuberculosis complex and it remains one of the leading causes of economic losses, morbidity and mortality throughout the world [1]. The dominant species of Mycobacterium tuberculosis complex (MTBC) are (M. tuberculosis, Mycobacterium Canetti, Mycobacterium africanum, Mycobacterium microti, M. bovis, Mycobacterium caprae and Mycobacterium pinnipedii) that are similar at genetic level [2]. M. tuberculosis is infecting more than one-third of the world’s human population and able to infect animals that have contact with humans. M. Canetti and M. africanum which are closely related to M. tuberculosis can also cause human TB. M. bovis have the broadest spectrum of host infection, affecting humans, domestic and wild Animals. The MTBC species are closely related taxonomic group of bacteria contributing to hundred percent chromosomal homology between each other and its genome is 4.4 Mbp and contains 0.01-0.3 per cent synonymic nucleotide polymorphisms [3]. Mycobacterium species that produce tuberculosis in humans and animals are merged in the MBTC and the mycobacteria grouped in the Mycobacterium tuberculosis complex is characterized by 99.9% similarity at the nucleotide level and identical 16S rRNA sequences but differ widely in terms of their host level tropisms, phenotypes, and pathogenicity.
The MTBC members have evolved from a common ancestor through deletion and insertion in the genome or DNA and causing the present mycobacterium speciation. Genomic analysis helps to identify regions of difference (RD) which is important to indicate chromosomal genes related to pathogenicity. M. tuberculosis is the most wide spread etiological agent of human tuberculosis evolved from M. bovis by specific adaptation of animal pathogen to the human host [4]. The members of MTBC are indistinguishable in their 16SrRNA and rpoB genes because the recombination events does not occur between strains rather their genomic length is the same, and the host specificity allows differentiation [5]. The large genome deletion named RDs which is present and absent in some strains in the mycobacterium tuberculosis complex are used for construction of molecular phylogeny due to their low mutation rate [6]. It is derived from a common ancestor which is intriguing that some are exclusively human (M. tuberculosis, Mycobacterium africanum, Mycobacterium Canetti) or rodent pathogens (Mycobacterium microti), whereas the others have a wide host spectrum (Mycobacterium bovis). MTBC has peculiar phylogenetic lineages due to its emerging genetic variation, host and environmental factors. The advancement of new molecular methods is used to understand the transmission dynamics and pathogenesis of the disease and genetic diversity of mycobacterium tuberculosis. The strains isolated from different hosts, geographic origins, and exhibits a wide spectrum characteristics are typed into groups of related strains through IS6110 restriction fragment length polymorphism [8], mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU VNTR) typing [10], Spoligotyping [9], targeting of large sequence polymorphisms (LSPs) (6) and typing of single nucleotide polymorphism [11].
The current typing of M. tuberculosis complex species is done by the restriction fragment length polymorphism (RFLP) -IS6110. LSPs and SNPs are considered be the most suitable markers for strain identification and phylogenetic analysis [12]. Spoligotyping is the most widely used method which allows the differentiation of strains inside each species belonging to M. tuberculosis complex, such as M. bovis from M. tuberculosis [9]. MIRU-VNTRs are based on PCR amplification of genetic elements that are located mainly in inter-genic regions dispersed throughout the MTB genome. Deletion of certain variable genomic regions did not occur separately in the different strains of M. tuberculosis complex whichallows a new scenario of the evolutionary process. There are a few reports on the molecular diversity of MTBC strains, only limited reports are available to determine the genetic diversity of MTBC strains circulating in the environment. The application of early diagnosis is highly important for the control of tuberculosis, rapid advancement methods in molecular strain typing would contribute significantly to understand the genetic diversity and circulation of mycobacterium strains as well as their distribution to establish the prevailing epidemiology and transmission pattern of the MBTC strains. Understanding and adopting the currently used molecular typing tool is useful. Therefore, the objectives of this work were:
- To highlight the molecular phylogeny and spread of mycobacterium tuberculosis complex strains
- To molecularly describe an alternative and advanced typing methods of MTBC strains that is adopted by producers
Molecular phylogeny of Mycobacterium Tuberculosis Complex
The large genome deletion named region of difference (RD) in the mycobacterium tuberculosis complex are used for construction of phylogenies due to their low mutation rate [7] because they are less likely to be homoplastic than SPN and are easy to analyze via PCR methods. Genetic sequence analysis of the RD deletions confirmed the same genetic events as the flanking sequences are similar for the isolates. In this regard RD 6 is exceptional because it is a highly repetitive region where its exact site is not confirmed [13]. The absence of RD6 represents the same large sequence polymorphism. Comparative genomics analysis shows that it is unlikely that M. tuberculosis evolved from M. bovis. The open reading frame structures at junction points indicated that RDS are deletions from the M. bovis genome rather than insertion into M. tuberculosis genome [14].
The large genome deletion named region of difference (RD) in the mycobacterium tuberculosis complex are used for construction of phylogenies due to their low mutation rate [7] because they are less likely to be homoplastic than SPN and are easy to analyze via PCR methods. Genetic sequence analysis of the RD deletions confirmed the same genetic events as the flanking sequences are similar for the isolates. In this regard RD 6 is exceptional because it is a highly repetitive region where its exact site is not confirmed [13]. The absence of RD6 represents the same large sequence polymorphism. Comparative genomics analysis shows that it is unlikely that M. tuberculosis evolved from M. bovis. The open reading frame structures at junction points indicated that RDS are deletions from the M. bovis genome rather than insertion into M. tuberculosis genome [14].
Insertion of a polygenic region into M. bovis genome causes the completion of existing truncated open reading frame which results in its disruption during the deletion event. Although selective pressure could lead to the convergence by deleting the same genomic region in unrelated strain and this is highly unlikely because independent deletion would not occur at the same base pair. The RDs are not flanked by repetitive sequences that are prone to generate identical deletions but the description of phylogeny of MBTC is based on the analysis of RDs and selection of SNP. The members of the M. tuberculosis complex are genetically similar, showing 99.9% similarity at nucleotide level and identical 16S rRNA sequences. But the split of the MTBC in different species reflects mostly host preference and host specific ecotypes that differ by their host adaptation [15]. Mycobacterium tuberculosis complex are derived from a common ancestor and some are exclusively human (M. tuberculosis, Mycobacterium africanum, Mycobacterium Canetti) or rodent pathogens (Mycobacterium microti), whereas others have a wide host spectrum such as M. bovis. The presence of high degree of conservation in the house keeping genes of strains, the members of M. tuberculosis complex underwent an evolutionary process at the time of speciation. It is the most widespread etiological agent of human tuberculosis which is evolved from M. bovis by specific adaptation of an animal pathogen to the human host [7]. The lineages inside M. tuberculosis could be envisioned as ecotypes as they show a geographic distribution preference which might be related to the human genotypes. Several variable genomic regions in the members of the M. tuberculosis complex and differential hybridization arrays identified 14 regions of difference (RD1–14), ranging from 2 to 12.7 kb, that is absent from bacillus chalmette Guerin Pasteur relative to M. tuberculosis H37Rv [9].
The strains isolated from different hosts and a broad range of geographic origins, exhibit a wide spectrum of typing characteristics like IS6110 and spoligotype hybridization patterns or variable number tandem repeats of mycobacterial interspersed repetitive unit , the deletion of certain variable genomic regions did not occur independently in the different strains of the M. tuberculosis complex. This allows completely new scenario for the evolution of the M. tuberculosis complex and the origin of human tuberculosis. The strains have been extensively characterized by reference typing methods, (IS6110 restriction fragment length polymorphism (RFLP) typing and spoligotyping. M. tuberculosis H37Rv, M. tuberculosis H37Ra, M. tuberculosis CDC1551, M. bovis AF212297, M. microti OV254, and M. Canetti CIPT 140010059 which are used as reference strains.
Occurrence of variable genomic regions in the species of mycobacterium complex
Genomic regions with altered synteny are identified in the virulent strains of M. bovis genome and there is also loss of gene synteny in few genomic regions. This is due to an evolutionary process of gene loss through deletions. A large genomic regions of a particular pathogen contain virulence genes that aid them in establishing infections. The detection of different strains within the MTBC depends on the analysis of phenotypic characteristics like colony morphology, growth rate, acid fast and biochemical tests. Fast species discrimination and understanding of the phylogenetic relations and the evolutionary origin of MTBC strain is easily performed by genotyping procedures. Virulent strain identification of the MTBC genotyping procedure is relied on the analysis of DNA of various strains. Bacillus Chalmette Guerin strains used oligonucleotides internal to know RDs and RvDs, as well as oligonucleotides flanking regions. This approach generated a large data set which is highly reliable, and internally controlled, because PCR amplicons obtained with the internal primer pair correlated with the absence of amplicon in the flanking primer-pair. According to the conservation of junction sequences, the flanking the variable regions are distinguished and each one have a different importance as an evolutionary marker. The first region includes mobile genetic elements, like the prophages phiRv1 (RD3) and phiRv2 (RD11) and insertion sequences IS1532 (RD6) and IS6110 (RD5), whose distribution in the tubercle bacilli is highly divergent. The other type of deletion which is mediated by recombination between adjacent IS6110 insertion elements results in the loss of the intervening DNA segment (RvD2, RvD3, RvD4, RvD5) which is variable from strain to strain. The deletions whose bordering genomic regions do not contain repetitive sequences occurred in coding regions resulting in the truncation of genes that are intact in other strains of the M. tuberculosis complex. The exact mechanism leading to this type of deletion remains unclear, but strand slippage errors of DNA polymerase may have contributed to this events. RD1, RD2, RD4, RD7, RD8, RD9, RD10, RD12, RD13, RD14, and TbD1 are representatives of this event whose distribution among the strains allow to proceed an evolutionary scenario for the members of the M. tuberculosis complex that identifies M. tuberculosis and or M. Canetti as most closely related to the common ancestor of the tubercle bacilli strain [6].
Genomic regions with altered synteny are identified in the virulent strains of M. bovis genome and there is also loss of gene synteny in few genomic regions. This is due to an evolutionary process of gene loss through deletions. A large genomic regions of a particular pathogen contain virulence genes that aid them in establishing infections. The detection of different strains within the MTBC depends on the analysis of phenotypic characteristics like colony morphology, growth rate, acid fast and biochemical tests. Fast species discrimination and understanding of the phylogenetic relations and the evolutionary origin of MTBC strain is easily performed by genotyping procedures. Virulent strain identification of the MTBC genotyping procedure is relied on the analysis of DNA of various strains. Bacillus Chalmette Guerin strains used oligonucleotides internal to know RDs and RvDs, as well as oligonucleotides flanking regions. This approach generated a large data set which is highly reliable, and internally controlled, because PCR amplicons obtained with the internal primer pair correlated with the absence of amplicon in the flanking primer-pair. According to the conservation of junction sequences, the flanking the variable regions are distinguished and each one have a different importance as an evolutionary marker. The first region includes mobile genetic elements, like the prophages phiRv1 (RD3) and phiRv2 (RD11) and insertion sequences IS1532 (RD6) and IS6110 (RD5), whose distribution in the tubercle bacilli is highly divergent. The other type of deletion which is mediated by recombination between adjacent IS6110 insertion elements results in the loss of the intervening DNA segment (RvD2, RvD3, RvD4, RvD5) which is variable from strain to strain. The deletions whose bordering genomic regions do not contain repetitive sequences occurred in coding regions resulting in the truncation of genes that are intact in other strains of the M. tuberculosis complex. The exact mechanism leading to this type of deletion remains unclear, but strand slippage errors of DNA polymerase may have contributed to this events. RD1, RD2, RD4, RD7, RD8, RD9, RD10, RD12, RD13, RD14, and TbD1 are representatives of this event whose distribution among the strains allow to proceed an evolutionary scenario for the members of the M. tuberculosis complex that identifies M. tuberculosis and or M. Canetti as most closely related to the common ancestor of the tubercle bacilli strain [6].
M. Tuberculosis strains
M. tuberculosis strains by deletion analysis contain RD regions. Region of difference 4 is a genetic segment that is deleted from M. bovis strain but present in other strains. Onlyregions RD3 and RD11, corresponding to the two prophagesphiRv1 and phiRv2 of M. tuberculosis H37Rv [16], RD6, containingthe insertion sequence IS1532, and RD5, that is flankedby a copy of IS6110 (Gordon et al., 1999), are absent in some strains. M. tuberculosis strains are highly conserved with respect to RD1, RD2, RD4, RD7, RD8, RD9, RD10, RD12, RD13, and RD14, and these RDs represent regions that can differentiate M. tuberculosis strains independently of their geographical origin and their typing characteristics from certain members of the M. tuberculosis complex. Based on the presence or absence of M. tuberculosis specific deletion (TbD1), M. tuberculosis strains can be divided into ancestral and modern strains. The modern type of strains represents African M. tuberculosis clusters. Furthermore, successive loss of DNA, reflected by region of difference nine and other subsequent deletions, was identified for an evolutionary lineage represented by M. africanum, M. microti, and M. bovis that diverged from the progenitor of the present M. tuberculosis strains before TbD1 occurred. M. tuberculosis strains where the TbD1 region is not deleted, contained a leucine (CTG) at katG463, which described as characteristic for ancestral M. tuberculosis strains. During the evolution of M. tuberculosis, the katG mutation at codon 463 CTG (Leu) 3CGG (Arg) occurred in a progenitor strain that had region TbD1 deleted. Strains belonging to group one may have deleted region TbD1, whereas strains belonging to groups 2 and 3 lacked TbD1. In addition, all strains of groups 2 and 3 characteristically lacked spacer sequences 33–36 in the direct repeat (DR) region. Hence, M. tuberculosis, evolved from M. bovis as the agent of bovine disease. M. Canetti and ancestral M. tuberculosis strains lack none of these deleted regions, and, therefore, seem to be direct descendants of tubercle bacilli that existed before the M. africanum. M. bovis lineage separated from the M. tuberculosis lineage. The common ancestor of the tubercle bacilli resembled M. tuberculosis or M. Canetti. TbD1-deleted strains will be referred to modern M. tuberculosis strains [6]
M. tuberculosis strains by deletion analysis contain RD regions. Region of difference 4 is a genetic segment that is deleted from M. bovis strain but present in other strains. Onlyregions RD3 and RD11, corresponding to the two prophagesphiRv1 and phiRv2 of M. tuberculosis H37Rv [16], RD6, containingthe insertion sequence IS1532, and RD5, that is flankedby a copy of IS6110 (Gordon et al., 1999), are absent in some strains. M. tuberculosis strains are highly conserved with respect to RD1, RD2, RD4, RD7, RD8, RD9, RD10, RD12, RD13, and RD14, and these RDs represent regions that can differentiate M. tuberculosis strains independently of their geographical origin and their typing characteristics from certain members of the M. tuberculosis complex. Based on the presence or absence of M. tuberculosis specific deletion (TbD1), M. tuberculosis strains can be divided into ancestral and modern strains. The modern type of strains represents African M. tuberculosis clusters. Furthermore, successive loss of DNA, reflected by region of difference nine and other subsequent deletions, was identified for an evolutionary lineage represented by M. africanum, M. microti, and M. bovis that diverged from the progenitor of the present M. tuberculosis strains before TbD1 occurred. M. tuberculosis strains where the TbD1 region is not deleted, contained a leucine (CTG) at katG463, which described as characteristic for ancestral M. tuberculosis strains. During the evolution of M. tuberculosis, the katG mutation at codon 463 CTG (Leu) 3CGG (Arg) occurred in a progenitor strain that had region TbD1 deleted. Strains belonging to group one may have deleted region TbD1, whereas strains belonging to groups 2 and 3 lacked TbD1. In addition, all strains of groups 2 and 3 characteristically lacked spacer sequences 33–36 in the direct repeat (DR) region. Hence, M. tuberculosis, evolved from M. bovis as the agent of bovine disease. M. Canetti and ancestral M. tuberculosis strains lack none of these deleted regions, and, therefore, seem to be direct descendants of tubercle bacilli that existed before the M. africanum. M. bovis lineage separated from the M. tuberculosis lineage. The common ancestor of the tubercle bacilli resembled M. tuberculosis or M. Canetti. TbD1-deleted strains will be referred to modern M. tuberculosis strains [6]
M. Canetti
M. Canetti is a very rare, smooth variant of M. tuberculosis and it shares identical 16S rRNA sequences with the other members of the M. tuberculosis complex, M. Canetti strains differ in polymorphisms in certain house-keeping genes, IS1081 copy number, colony morphology, and the lipid content of the cell wall [17]. In M. Canetti, the RD, RvD, and TbD1 regions except the prophages (phiRv1, phiRv2) are present. RD can be absent from M. Canetti strains that partially overlapped RD12. The conservation of RD, RvD, and TbD1 regions in the genome of M. Canetti indicated that M. Canetti diverged from the common ancestor of the M. tuberculosis complex before RD, RvD, and TbD1 occurred in the lineages of tubercle bacilli. Itwas shown to carry 26 unique spacer sequences in the DR region [18] that are not present in any other member of the M. tuberculosis complex. Therefore, M. Canetti represents a tubercle bacillus, whose genomic analysis reveals the evolution of the M. tuberculosis complex.
M. Canetti is a very rare, smooth variant of M. tuberculosis and it shares identical 16S rRNA sequences with the other members of the M. tuberculosis complex, M. Canetti strains differ in polymorphisms in certain house-keeping genes, IS1081 copy number, colony morphology, and the lipid content of the cell wall [17]. In M. Canetti, the RD, RvD, and TbD1 regions except the prophages (phiRv1, phiRv2) are present. RD can be absent from M. Canetti strains that partially overlapped RD12. The conservation of RD, RvD, and TbD1 regions in the genome of M. Canetti indicated that M. Canetti diverged from the common ancestor of the M. tuberculosis complex before RD, RvD, and TbD1 occurred in the lineages of tubercle bacilli. Itwas shown to carry 26 unique spacer sequences in the DR region [18] that are not present in any other member of the M. tuberculosis complex. Therefore, M. Canetti represents a tubercle bacillus, whose genomic analysis reveals the evolution of the M. tuberculosis complex.
M. Africanum
This isolates originate from West and East African sources and the junction sequences bordering RD7, RD8, and RD10, like those for RD9, are identical to those of M. bovis and M. microti strains. Two prophages phiRv1 and phiRv2, the West African strains contained phiRv2, whereas phiRv1 is absent. RvD1-RvD5 and TbD1 regions are present in these strains. M. africanum be differentiated from modern M. tuberculosis by the absence of region RD9 and the presence of region TbD1 variable genetic markers. The absence of the TbD1 region is confirmed by sequence analysis of the TbD1 junction region, which is found to be identical to that of TbD1-deleted M. tuberculosis strains. This indicates a very close genetic relationship of these strains to M. tuberculosis and it is regarded as M. tuberculosis rather than M. africanum strains [6].
This isolates originate from West and East African sources and the junction sequences bordering RD7, RD8, and RD10, like those for RD9, are identical to those of M. bovis and M. microti strains. Two prophages phiRv1 and phiRv2, the West African strains contained phiRv2, whereas phiRv1 is absent. RvD1-RvD5 and TbD1 regions are present in these strains. M. africanum be differentiated from modern M. tuberculosis by the absence of region RD9 and the presence of region TbD1 variable genetic markers. The absence of the TbD1 region is confirmed by sequence analysis of the TbD1 junction region, which is found to be identical to that of TbD1-deleted M. tuberculosis strains. This indicates a very close genetic relationship of these strains to M. tuberculosis and it is regarded as M. tuberculosis rather than M. africanum strains [6].
M. Bovis Comprises oflarge host spectrum infecting mammalian species, including man. M. bovis is the final member of a separate lineage represented by M. africanum (RD9), M. microti (RD7, RD8, RD9, RD10) and M. bovis (RD4, RD5, RD7, RD8, RD9, RD10, RD12, RD13) that branched from the progenitor of M. tuberculosis isolates. Successive loss of DNA have contributed to clonal expansion and the appearance of more successful pathogens in certain new hosts. The conservation of the TbD1 junction sequence in tested TbD1 deleted strains indicated it is, a descent from a single clone. Therefore, the TbD1 deletion is a good indicator that modern M. tuberculosis strains account for the vast majority of today’s tuberculosis cases undergone this evolutionary processes and spread around the world. The other major bottleneck, which seems to have occurred for members of the M. africanum and M. microti. M. bovis lineage is reflected by RD9 and the subsequent RD7, RD8, and RD10 deletions. These deletions have occurred in the progenitor of tubercle bacilli that shows natural host spectra as diverse as humans in Africa. The Mycobacterium whose DNA is amplified showed a spoligotype thatis most closely related to patterns of M. africanum and couldhave been an early representative of the lineage M. africanum 3 M. bovis.
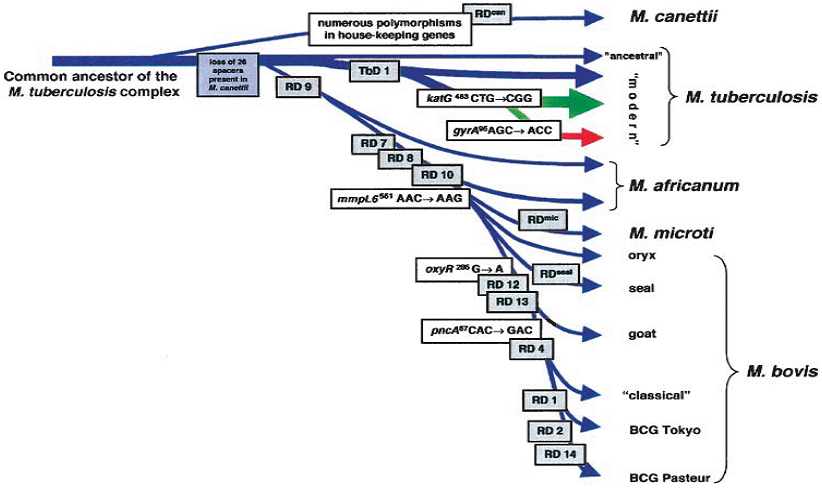
Figure 1: The evolutionary pathway of MBTC based on the presence and absence of regions of difference and SNPS, [6].
Advanced Molecular methods for diagnosis and typing of MBTC strains
Advanced molecular methods are commonly used to trace strains of MTBC and their epidemiological distribution. Advances in molecular techniques allows to distinguish the genus mycobacterium using sensitive and rapid techniques based on the amplification of nucleic acid sequences from 16s ribosomal ribonucleic acid 16s rRNA [19]. Ribosomal RNA is an essential constituent of bacterial ribosome that contains highly conserved sequences and some variability. The M. tuberculosis genome is homogeneous with 99.9% similarity and identity to the 16s rRNA sequence, with M. africanum and M. mungi being species genetically related to the M. tuberculosis complex and undergoing mutations. The reference molecular method for identification of mycobacteria is the determination of sequences of 16S ribosomal DNA, the molecule is highly conserved. The 16s rRNA is 1500 nucleotide in length and a component of the smaller subunit of the 70s prokaryotic ribosome whereas the small but the conserved sequence difference in the rRNA molecule which helps to differentiate at genus, group or species level [20].
Advanced molecular methods are commonly used to trace strains of MTBC and their epidemiological distribution. Advances in molecular techniques allows to distinguish the genus mycobacterium using sensitive and rapid techniques based on the amplification of nucleic acid sequences from 16s ribosomal ribonucleic acid 16s rRNA [19]. Ribosomal RNA is an essential constituent of bacterial ribosome that contains highly conserved sequences and some variability. The M. tuberculosis genome is homogeneous with 99.9% similarity and identity to the 16s rRNA sequence, with M. africanum and M. mungi being species genetically related to the M. tuberculosis complex and undergoing mutations. The reference molecular method for identification of mycobacteria is the determination of sequences of 16S ribosomal DNA, the molecule is highly conserved. The 16s rRNA is 1500 nucleotide in length and a component of the smaller subunit of the 70s prokaryotic ribosome whereas the small but the conserved sequence difference in the rRNA molecule which helps to differentiate at genus, group or species level [20].
Molecular Strain typing methods based on genomic DNA
Restriction fragment length polymorphism Is6110 Typing
The use of IS6110 is the most frequently used method for isolates of members of the M. tuberculosis complex. The RFLP IS6110 technique is labor-intensive, slow and requiring a large quantity of good-quality DNA (1-2 µg), But it is limited to discriminate isolates with less than 6 copies of IS6110 (< 6 bands in the RFLP pattern) and difficulty in comparing results obtained in different laboratories. This molecular marker contains stability of one to three years in patterns of RFLP IS6110, which is related to the number of copies present, as a larger number of copies imply a greater possibility of transposition among isolates of 8 to 12 copies [21].
Restriction fragment length polymorphism Is6110 Typing
The use of IS6110 is the most frequently used method for isolates of members of the M. tuberculosis complex. The RFLP IS6110 technique is labor-intensive, slow and requiring a large quantity of good-quality DNA (1-2 µg), But it is limited to discriminate isolates with less than 6 copies of IS6110 (< 6 bands in the RFLP pattern) and difficulty in comparing results obtained in different laboratories. This molecular marker contains stability of one to three years in patterns of RFLP IS6110, which is related to the number of copies present, as a larger number of copies imply a greater possibility of transposition among isolates of 8 to 12 copies [21].
Polymorphic Guanine-cytosine rich sequence Analysis (PGRS)
This method is useful in typing clinical isolates of members of the M. tuberculosis complex that contains a low number of copies of the insertion sequence IS6110. These repetitive sequences are present in multiple copies through the mycobacterium genome; 26 to 30 copies of repetitive sequences are found per chromosome in members of the M. tuberculosis complex. It has a discriminating power very similar to that of RFLP, requiring DNA of good quality. It is limited to discriminate isolates that possess multiple copies of this genetic marker [22].
This method is useful in typing clinical isolates of members of the M. tuberculosis complex that contains a low number of copies of the insertion sequence IS6110. These repetitive sequences are present in multiple copies through the mycobacterium genome; 26 to 30 copies of repetitive sequences are found per chromosome in members of the M. tuberculosis complex. It has a discriminating power very similar to that of RFLP, requiring DNA of good quality. It is limited to discriminate isolates that possess multiple copies of this genetic marker [22].
Analysis and pulsed-field gel electrophoresis (PFGE)
This method possesses a high power of discrimination, requiring large concentrations of DNA with purity and quality. The introduction of PFGE has had great impact in the study of mycobacterial genome and genomic maps of closely related species such as those that comprise the M. tuberculosis complex, thus allowing for the establishment of differences through the identification of multiple rearrangements and the non-random location of insertion elements [23].
This method possesses a high power of discrimination, requiring large concentrations of DNA with purity and quality. The introduction of PFGE has had great impact in the study of mycobacterial genome and genomic maps of closely related species such as those that comprise the M. tuberculosis complex, thus allowing for the establishment of differences through the identification of multiple rearrangements and the non-random location of insertion elements [23].
Molecular typing methods based on PCR product
Spoligotyping (spacer oligonucleotide genotyping)
The direct repetitive regions of the direct repeat sequence in members of the strains of M. tuberculosis complex are composed of multiple direct variant repetitive sequences, each of which is comprised of direct repetitions of 36 pairs of bases separated by unique spacer sequences of 35 to 41 pb, generating a large polymorphism which can be used in molecular epidemiological studies for the differentiation of species of the M. tuberculosis complex. Spoligotyping is based on PCR which is targeted on a small DR sequence within a spacer region. The presence or absence and deletion of spologotye spacers or patterns of these sequences is determined by hybridization with a set of 43 oligonucleotides derived from M. tuberculosis H37Rv. This technique is useful in the typing of clinical isolates of species of the M. tuberculosis complex, in those with less than 6 copies of IS6110, and in different clinical samples [24].
Spoligotyping (spacer oligonucleotide genotyping)
The direct repetitive regions of the direct repeat sequence in members of the strains of M. tuberculosis complex are composed of multiple direct variant repetitive sequences, each of which is comprised of direct repetitions of 36 pairs of bases separated by unique spacer sequences of 35 to 41 pb, generating a large polymorphism which can be used in molecular epidemiological studies for the differentiation of species of the M. tuberculosis complex. Spoligotyping is based on PCR which is targeted on a small DR sequence within a spacer region. The presence or absence and deletion of spologotye spacers or patterns of these sequences is determined by hybridization with a set of 43 oligonucleotides derived from M. tuberculosis H37Rv. This technique is useful in the typing of clinical isolates of species of the M. tuberculosis complex, in those with less than 6 copies of IS6110, and in different clinical samples [24].
This method identifies polymorphism in the presence of spacer units in the direct-repeat (DR) region in strains of the M. tuberculosis complex. The DR is composed of multiple, identical 36-bp regions interspersed with unique DNA spacer sequences of similar size. Spacer sequences are unique to the DR region, and copies are not located in the chromosome. The spoligotyping assay exploits polymorphisms in spacer sequences found in the direct repeat locus in the chromosome of MTC strains [9]. The locus consists of well-conserved direct repeats interspersed with unique spacer sequences. The region comprising the repeat plus the adjacent spacer has been termed the direct variable repeat. Spoligotyping is fast, highly reproducible method, and the genotyping result has a simple binary format, which permits the exchange of data and facilitates the construction of large collaborative databases [25]. Spoligotyping differentiates isolates of MTC strains by determining the absence or presence of the 43 defined spacer sequences. The order of the spacers was found to be well conserved [8]. Spoligotyping detects and identifies MTBC without requiring purified DNA which is based on the polymorphism direct repeat locus of variable repeats. It can be applied to cultured cells to determine the presence or absence of spacers. Hence, spoligotyping can show the strain is circulating in the cattle population clustered in different strains show signal RD deletions. Spoligotyping is therefore depends on the variability of spacer sequences interspersed with repeat sequences in the direct repeat locus. Strains vary in the number of DRs and presence or absence of spoligotype spacers.
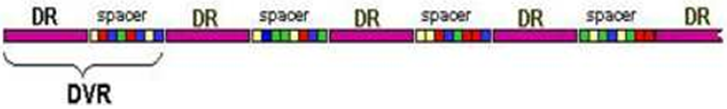
Figure 2: Direct locus [8].
Fast ligation mediated PCR (Flip)
A rapid method with high reproducibility and great power of discrimination, based on the study of the IS6110 sequence. In comparison with the RFLP IS6110 method, this technique requires small quantities of DNA or crude cell lysates. It has limited discriminating power in isolates with less than six copies of the IS6110 sequence and gives results in less time than LM-PCR method [26].
A rapid method with high reproducibility and great power of discrimination, based on the study of the IS6110 sequence. In comparison with the RFLP IS6110 method, this technique requires small quantities of DNA or crude cell lysates. It has limited discriminating power in isolates with less than six copies of the IS6110 sequence and gives results in less time than LM-PCR method [26].
Double Repetitive Element PCR technique (DRE-PCR)
A methodology with great power of discrimination that consists in the amplification of DNA segments located between the IS6110 sequence and the polymorphic region rich in guanine cytosine. This method is based on the number of copies and the distance between the repetitive sequences of IS6110. The distances vary among the clinical isolates and the variations allow differentiation with respect to the size and number of amplified fragments of DNA, producing a single pattern band for the different strains of M. tuberculosis. It requires primary culture. The resolution power of this method is poo [27].
A methodology with great power of discrimination that consists in the amplification of DNA segments located between the IS6110 sequence and the polymorphic region rich in guanine cytosine. This method is based on the number of copies and the distance between the repetitive sequences of IS6110. The distances vary among the clinical isolates and the variations allow differentiation with respect to the size and number of amplified fragments of DNA, producing a single pattern band for the different strains of M. tuberculosis. It requires primary culture. The resolution power of this method is poo [27].
Mycobacterial interspersed repetitive units-variable number of tandem repeats (MIRU-VNTR)
It requires short tandem repeat structures found at multiple loci throughout the M. Tuberculosis genome and have been used for typing these pathogens. It allows splitting certain spoligotyping defined clusters in smaller sub clusters. This method of typing M. tuberculosis complex is based on the variable number tandem repeat or MIRU, with a power of resolution similar to that produced by RFLP IS6110. The MIRU are short elements of DNA (40 to 100 pb) found in tandem repetitions and dispersed in inter-genic regions of the genome within members of the complex. The clinical isolates characterized by these genetic markers are represented with a 12- digit code corresponding to the number of repetitions in each MIRU locus, there by forming the basis of a system that facilitates global comparison among laboratories [28].
It requires short tandem repeat structures found at multiple loci throughout the M. Tuberculosis genome and have been used for typing these pathogens. It allows splitting certain spoligotyping defined clusters in smaller sub clusters. This method of typing M. tuberculosis complex is based on the variable number tandem repeat or MIRU, with a power of resolution similar to that produced by RFLP IS6110. The MIRU are short elements of DNA (40 to 100 pb) found in tandem repetitions and dispersed in inter-genic regions of the genome within members of the complex. The clinical isolates characterized by these genetic markers are represented with a 12- digit code corresponding to the number of repetitions in each MIRU locus, there by forming the basis of a system that facilitates global comparison among laboratories [28].
Ligation Mediated PCR (LM-PCR)
This is a highly reproducible technique that uses a primer directed towards a specific region of insertion sequence IS6110, and targeted to a linker ligated to restricted genomic DNA. It requires small quantities of DNA and has a high power of discrimination [29].
This is a highly reproducible technique that uses a primer directed towards a specific region of insertion sequence IS6110, and targeted to a linker ligated to restricted genomic DNA. It requires small quantities of DNA and has a high power of discrimination [29].
Fluorescent Amplified Fragment Length Polymorphism (FAFLP)
This method is complementary technique for the typing of M. tuberculosis, in clinical isolates with less than 6 copies of IS6110. It requires the extracted DNA which is digested with EcoRI and Msel restriction enzymes [30].
This method is complementary technique for the typing of M. tuberculosis, in clinical isolates with less than 6 copies of IS6110. It requires the extracted DNA which is digested with EcoRI and Msel restriction enzymes [30].
Single Nucleotide Polymorphism Typing (SNP)
SNPs within miRNAs can change their characteristics via altering their target selection or expression, resulting in functional and phenotypic changes. The result obtained from this method is similar to those obtained from RFLP IS6110 and spoligotyping [31].
SNPs within miRNAs can change their characteristics via altering their target selection or expression, resulting in functional and phenotypic changes. The result obtained from this method is similar to those obtained from RFLP IS6110 and spoligotyping [31].
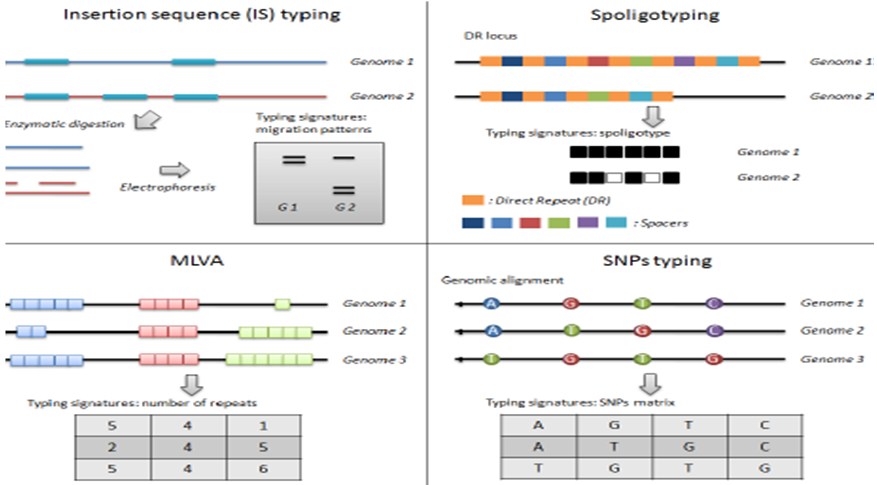
Figure 3: The principle of major genotyping techniques [32].
Multiplex polymerase chain Reaction
mPCR is a rapid diagnostic method which is used to differentiate MTBC strains and other mycobacterial species. The genomic DNA is used as sampling template and The PCR product is used for sequencing procedures.
mPCR is a rapid diagnostic method which is used to differentiate MTBC strains and other mycobacterial species. The genomic DNA is used as sampling template and The PCR product is used for sequencing procedures.
Spread of the mycobacterium tuberculosis complex
The modern genetic research indicates that domestication of European cattle took place in the near east at the beginning of Neolithic [33]. Studies based on mitochondrial DNA of cattle origin allow the expansion of cattle from an ancestral population from the Fertile Crescent [34]. The introduction of cattle into Europe occurred along with human migration following land routes but maritime routes which is reflected in the influence of cattle of North African origin in the Mediterranean countries and the same scenario is probably valid for goat population since cattle and goats are associated with mankind. Infected cattle and goat population gave rise to locally adapted strains and this evolutionary scenario is congruent with the demography of MBTC and its association with the human host [35]. The pathogen used the ecological niche which led to the clonal expansion of these strains. This is coherent with the hypothesis of ancient cattle being infected with ancestral M. bovis strains which is present at a maximum number of spacers in the DR region. These ancestors derived from M. tuberculosis like organism and generated strains that become adapted to different hosts [6].
The modern genetic research indicates that domestication of European cattle took place in the near east at the beginning of Neolithic [33]. Studies based on mitochondrial DNA of cattle origin allow the expansion of cattle from an ancestral population from the Fertile Crescent [34]. The introduction of cattle into Europe occurred along with human migration following land routes but maritime routes which is reflected in the influence of cattle of North African origin in the Mediterranean countries and the same scenario is probably valid for goat population since cattle and goats are associated with mankind. Infected cattle and goat population gave rise to locally adapted strains and this evolutionary scenario is congruent with the demography of MBTC and its association with the human host [35]. The pathogen used the ecological niche which led to the clonal expansion of these strains. This is coherent with the hypothesis of ancient cattle being infected with ancestral M. bovis strains which is present at a maximum number of spacers in the DR region. These ancestors derived from M. tuberculosis like organism and generated strains that become adapted to different hosts [6].
Control and prevention Measures
The effective control and eradication of BTB from herds or farms of cattle depend on identifying and isolating potential sources of infection through test-and-slaughter-strategies. There are various methods of eradication and control options adopted in different countries. In developed countries, BTB has nearly been reduced in farm through test-and-slaughter methods [36]. In developing countries, these measures cannot be adopted in practice due to financial constraint, scarcity of trained professionals, traditional beliefs, geographical barriers and less attention given to the importance of zoonotic tuberculosis in both animals and humans. The elimination of bovine tuberculosis has yet not been achieved due to the existence of wildlife reservoirs of m. bovis infection and poor knowledge about wildlife tuberculosis [37]. BTB can be controlled by adopting test and isolation of reactors, pasteurization of milk prior to consumption or further processing practices, culling of infected animals and improving sanitary and hygienic practices. Potent development of effective vaccines against MBTC strain is also recommended.
The effective control and eradication of BTB from herds or farms of cattle depend on identifying and isolating potential sources of infection through test-and-slaughter-strategies. There are various methods of eradication and control options adopted in different countries. In developed countries, BTB has nearly been reduced in farm through test-and-slaughter methods [36]. In developing countries, these measures cannot be adopted in practice due to financial constraint, scarcity of trained professionals, traditional beliefs, geographical barriers and less attention given to the importance of zoonotic tuberculosis in both animals and humans. The elimination of bovine tuberculosis has yet not been achieved due to the existence of wildlife reservoirs of m. bovis infection and poor knowledge about wildlife tuberculosis [37]. BTB can be controlled by adopting test and isolation of reactors, pasteurization of milk prior to consumption or further processing practices, culling of infected animals and improving sanitary and hygienic practices. Potent development of effective vaccines against MBTC strain is also recommended.
Concussion and recommendations
Mycobacterium species that produce tuberculosis in humans and animals are merged in the MBTC and characterized by 99.9% similarity at the nucleotide level and identical 16S rRNA sequences but differ widely in terms of host tropisms, phenotypes, and pathogenicity. MTBC members have evolved from a common ancestor through successive DNA deletions and insertions. The genomic analysis helps to identify regions of difference (RD) which is important to pinpoint chromosomal genes related to pathogenicity. The advancement of recent molecular techniques greatly contributed and enhanced to understand the transmission dynamics of MTBC within population and between hosts. Several methods are used to classify MTBC isolates into groups of related strains level through IS6110 restriction fragment length polymorphism, mycobacterial interspersed repetitive unit-variable number tandem repeat (MIRU-VNTR) typing based on PCR amplification of genetic elements that are located mainly in intergenic regions dispersed throughout the MTB genome and spoligotyping. As early diagnosis is important to control tuberculosis, recent advancement methods in molecular strain typing would contribute significantly to understand the genetic diversity and circulation of mycobacterium strains in animals. Therefore, further research in identifying the circulating strains of MBTC in various hosts and their distribution in different geographical area should be undertaken, molecular and genetic mapping of the isolates of MBTC in terms of their spatial and temporal distribution is strongly encouraged, Regular tuberculin skin testing and abattoir surveillance should be done in order to upgrade the knowledge and attitude of livestock keeping communities.
References
- WHO. (2013). Global tuberculosis report 2013,WHO/HTM/TB/2013.11,WHO press, Geneva, Switzerland.
- Pal, M. and Boru, B. (2012). Zoonotic significance of M. bovis infection. J. Natt. Hist. 8:86 -89.
- Sujatha, N. (2015): Genetic markers, genotyping methods and next gene ration sequencing in mycobacterium tuberculosis, Indian J Med. Res. 141: Pp.761-774.
- Stead, W. W., Eisenach, K. D., G. L., Thoen, C. O. and Bates, J. H. (1995). Am. J. Respir. Crit. Care Med. 151: 1267–1268.
- Garcia, B., J.C., Menendez, M., P., Garcia, M. (2011). Alignment of multiple complete genomes and gene rearrangements towards the speciation of mycobacteria. Infect Genet Evol. 12: 819–826.
- Gagneux S. (2006). Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA. 103: 2869– 2873.
- Brosch, S.V., Gordon, M., Marmiesse, P, Brodin. , C, Buchrieser, and K, Eiglmeier. (2002). A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Nat Acad Sci USA, 99: 3684–3689.
- Van Embden, J.A., van Gorkom, T., Kremer, K., Jansen, R. (2000). Genetic variation and evolutionary origin of the direct repeat locus of Mycobacterium tuberculosis complex bacteria. J. Bacteriol. 182: 2393–2401.
- Kamerbeek., J, Schouls., L, Kolk., A, van Agterveld M, van Soolingen D, Kuijper S, (1997). Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J Clin Microbiol. 35: 907–14.
- Supply Pet. (2006). Proposal for standardization of mycobacterial interspersed repetitive unit variable number tandem repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 44: 4498– 4510.
- Filliol, I. (2006). Global phylogeny of Mycobacterium tuberculosis based on single nucleotide polymorphism analysis, tuberculosis evolution, phylogenetic accuracy of other DNA, and recommendations for a minimal standard SNP set. J. Bacteriol. 188: 759– 772.
- Nakanishi, N., Wada, T., Arikawa, K., Millet, J., Rastogi., N, Iwamoto., T. (2013). Evolutionary robust SNPs reveal the misclassification of Mycobacterium tuberculosis Beijing family strains into sub lineages. Infect. Genet. Evol. 16: 174–177.
- Mostowy, S., Cousins, D., J. Brinkman, A., Aranza and M. Ber. (2002). Genomic deletions suggest bacillus Phylogeny for the MBTC. J. infect Dis 186: 74-80.
- Gordon, S. V., Brosch, R., Garnier, K. and Cole, S. T. (1999). Mol. Microbiol. 32: 643–655.
- Smith, N. (2006). A re -evaluation of M. proto tuberculosis PloS Pathos, Page 98.
- Cole, S.T., Brosch, R., Parkhill, J., Garnier, T., and Barrel, B.G. (1998). Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 393: 537-44.
- Van Soolingen, D., Hoogenboezem, T., Teppema, K. S., Brennan, P. J. (1997). Int. J. Syst. Bacteriol. 47: 1236–1245.
- Van-embden. (2000). Strain identification of Mycobacterium tuberculosis by DNA fingerprinting, J. Clin. Microbiol. 31: 406– 409.
- Rogall., T.J, Woters, T. Flohr and E.C Botter. (1990). Towards Phylogeny and definition of species at the molecular level within the genus mycobacterium. int.J.syst.Bacteriol. 40: 323-330.
- Tortil, E. (2003). Impact of genotypic studies on bacterial taxonomy the new mycobacterium of the 1990s. clin.micobio.Rev. 16: 319-354.
- Gopaul. , K, Brown and Gibson L. (2006). Progression toward an improved DNA amplification-based typing technique in the study of Mycobacterium tuberculosis epidemiology. J Clin Microbiol. 44: 2492-2498.
- Rozo. , A, Ribón. C. and Wellman. (2010). Molecular tools for Mycobacterium tuberculosis genotyping. Rev. salud Publica. 12 (3): 510-521.
- Wolfgan. P. Gordon., S. telenti., A. and Cole S. (1998). Pulsed field Gel Electrophoresis for Mycobacteria. In: Mycobacteria Protocols. Tanya Paris, New Jersey Pre .Pp.51- 63.
- Doroudchi. M, Kremer., K and E. Basiri. (2000). IS6110-RFLP and Spoligotyping of Mycobacterium tuberculosis isolates in Iran. Scand J Infect Dis. 32: 663-668.
- Filliol, I., Driscoll, J.R., D., Bifani, P.J., Brudey, K., J., C., Rastogi, N. (2003). Snapshot of moving and expanding clones of Mycobacterium tuberculosis and their global distribution assessed by spoligotyping in an international study. J. Clin. Microbiol. 41: 1963–1970.
- Reisig, F., Kremer. K, Amthor B, and Haas W. (2005). Fast Ligation Mediated PCR a Fast and Reliable Method for IS6110 Base Typing of Mycobacterium tuberculosis Complex. Journal of Clinical Microbiology. 43(11): 5622- 27.
- Mostron., P, Gordon., M, Sola C, Ridell M, Rastogui N. (2002). Methods used in the Molecular epidemiology of tuberculosis. Clin Microbiol Infect. 8:694-704.
- Narayanan, S. (2004). Molecular epidemiology of tuberculosis. Indian J Med Res. 120:233-247.
- Kremer, K., Arnold, C., Cataldi, A., Gutierrez, M., Haas W., Supply, D. (2005). Discriminatory power and reproducibility of novel DNA typing methods for Mycobacterium tuberculosis complex strains. J Clin Microbiol. 43: 5628-5638.
- Mortimer, P., Arnold, C. (2001). FAFLP last word in microbial genotyping, J Med Microbiol. 50: 393-395.
- Lid Wang, T, Song X, Yang B, Wang J, Ying B and Tao C. (2011). Genetic study of two single nucleotide polymorphisms within corresponding microRNAs and susceptibility to tuberculosis in a Chinese Tibetan and Han population. Human Immunology. 72: 598-602.
- Yann, B. (2014). A new scenario for the early evolution of mycobacterium tuberculosis. Bacteriology, university Paris.
- Edward, C., J., Bollongino, Larson, s .y, Bradley. andJ.Burger. (2007). Mitochondrial DNA analysis shows a Near eastern Neolithic origin for domestic cattle and no indication of domestication of European aurochs. proch, Biol sci. 274: 1377-1385.
- Beja, P., D. CaramelliN. Ferrand., F. Goyache and G. Bertorelle. (2006). The origin of European cattle evidence from modern and ancient DNA proc Natl, Acad, scie. U.S.A. 103: 8113-8118
- Hershberg, R., M, Lipatov, C.Roach,k. kremer, D. A.petrov, M.W. Feldma and Gangnex. (2008). high functional diversity in mycobacterium tuberculosis driven by genetic shift and human demography. plos Biol 6.
- Pavlik I., Ayele, W.,Y, Parmova I, Melicharek I, Hanzlikova M, Kormendy B., Nagy G., Cvetnic Z, Ocepek M, Fejzic N, Lipiec M. (2002). Incidence of bovine tuberculosis in cattle in seven Central European countries during the years 1990–1999. Veterinary Medicine. 47: 45–51.
- Mairtin, D., O., Williams D, H, Dolan L, and Collins J.D. (1998). The influence of selected herd factors and a badger-intervention tuberculosis-control Programme on the risk of a herd-level trade restriction to a bovine population in Ireland. Prev. vet. Med. 35: 79-90.
Citation: Beksisa Urge Hurrisa. (2020). New Evolutionary Scenario and Molecular Diagnostic Methods in typing of Mycobacterium Tuberculosis Complex strains. Archives of Veterinary and Animal Sciences 2(1). DOI: 10.5281/zenodo.3812681
Copyright: © 2020 Beksisa Urge Hurrisa. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.