Research Article
Volume 1 Issue 2 - 2019
Effect of Certain Polymeric Resins as Well as Water Soluble Carriers on the Release of Aceclofenac and its Ulcerogenic Activity in Rats
1Pharm. Technology Dept., Faculty of Pharmacy, Tanta University*, Tanta, Egypt
2Pharmaceutics Dep. Faculty of Pharmacy, Egyptian Russian University, Cairo, Egypt
2Pharmaceutics Dep. Faculty of Pharmacy, Egyptian Russian University, Cairo, Egypt
*Corresponding Author: Esmat E. Zein, Pharm. Technology Dept., Faculty of Pharmacy, Tanta University, Tanta, Egypt.
Received: November 07, 2019; Published: November 18, 2019
Abstract
Adverse effects of non-steroidal anti-inflammatory drugs (NSAIDs) are seen in almost all organs but GI tract is most frequently affected as they cause GI irritation, bleeding and also peptic as well as duodenal ulceration. Concepts about gastro-duodenal mucosal ulceration have been evolved from the simple theory of topical injury to other theories involving multiple mechanisms with both local and systemic effects. Aceclofenac is one of the commonly used NSAIDs. It belongs to class II drugs in BCS and characterized by poor aqueous solubility and high absorption. Dissolution of aceclofenac is a rate limiting step which affects its onset especially in dental pain, rheumatoid and osteoarthritis. Solid dispersion is a type of solid state material where molecular dispersion of one or more active drugs is carried by an inert carrier. The above mentioned considerations led to the objective of this study, which aimed to prepare and evaluate solid dispersion systems containing aceclofenac and Eudragit Ll00, Eudragit S100, Eudragit RL100, PVP k90 and methyl cellulose 15cps. All formulations were prepared by dissolving the polymer in a mixture of isopropanol and acetone (1:1 V/V), 100mg drug was then dissolved in a minimal amount of the solvents mixture at 40oC. Solvent were evaporated over a period of 24 hrs under stirring conditions (150 rpm) at room temperature, 1:1. 1:2 and 1:3 ratios were utilized for each polymer. The polymer which showed the optimum release conditions was chosen for in-vivo studies using male Wistar rats. The obtained solid dispersion systems were evaluated using FT-IR, DSC and PXRD. Drug content as well as in- vitro drug release studies were determined at different pH values. Drug content in the prepared matrices ranged between 97.98% and 100%. No significant drug-polymer interactions were observed in IR studies. Aceclofenac entrapment efficiency in the different prepared formulations was affected by neither the polymer type nor drug to polymer ratio. Solid dispersion with Eud S100 1:1 drug to polymer significantly reduced gastric irritations and gastric ulcers compared to free drug as well as the other proposed formulations as proved from histopathological examination of rat stomachs.
Key words: Solid dispersion; Aceclofenac; NSAIDs; Drug delivery systems; Ulcerogenic activity
Introduction
Aceclofenac (2-[2-[2-[(2,6-dichlorophenyl)amino]- phenyl]acetyl] oxyacetic acid) (Figure 1), is a NSAID used in the first line treatment of osteoarthritis, rheumatoid arthritis and ankylosing spondylitis [1].
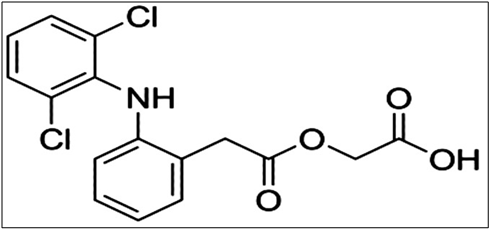
Figure 1: Structure of aceclofenac.
Aceclofenac is a partially water insoluble drug which undergoes first pass metabolism when taken orally. The drug is well absorbed after oral administration with hepatic first-pass metabolism [2]. Aceclofenac belongs to class II drugs in BCS characterized by poor aqueous solubility and high absorption, dissolution of aceclofenac is rate limiting step that affects its onset which is important in some cases such as dental pain, rheumatoid and osteoarthritis.
Concepts about gastro-duodenal mucosal injury induced by NSAIDs have been evolved from the simple theory of topical injury to other theories involving multiple mechanisms with both local and systemic effects [3, 4]. Topical mucosal injury of NSAIDs is initiated by their acidic properties represented by their lower dissociation constant. These weak acids remain as lipophilic non-ionized form in the highly acidic gastric environment such conditions favor their migration through mucus across plasma membranes into surface epithelial cells, where NSAIDs are dissociated resulting in trapping of hydrogen ions that can directly kill epithelial cells [5-7]. NSAIDs can also induce topical mucosal damage by decreasing the hydrophobicity of gastric mucosa, thereby allowing endogenous gastric acid and pepsin to injure and damage the surface epithelium [8, 9]. Although NSAIDs had a well-established position in the management of osteoarthritis and rheumatoid arthritis, its chronic use is accompanied with significant gastrointestinal toxicity [10]. Patients chronically taking these drugs clinically develop significant ulceration, bleeding and obstruction [11]. The highest rate of upper gastrointestinal bleeding was reported for aceclofenac users among different users of other NSAIDs. Gastrointestinal adhesion of aceclofenac is directly related to its cumulative retention inside the GIT.
Previous investigations were reported which aimed to overcome the aforementioned problems by formulating soft capsules containing drug and solubilizers [12], solid dispersion using surfactants [13], complexation with HP-β-cyclodextrin [14], dual release compositions of cox-2 inhibitors [15], spherical agglomerates using sodium alginate and PVP [16], chitosan drug co-crystals [1], drug-loaded agarose beads [17], enteric coated immediate release pellets [18] as well as fast dissolving tablets [19]. All mentioned approaches seemed to be more efficient for improving the dissolution of these drugs rather than avoiding their GI adverse effects.
It was found that there is a critical need to prepare a suitable formula for aceclofenac not only to improve its solubility and physicochemical properties but also to minimize its GI side effects. Different microencapsulation techniques were adopted for this purpose [20-24], where they act by encapsulating the drug by polymeric coat which decreases the direct contact between the drug dispersed inside the polymeric matrix and the gastric mucosa. On the other hand protective polymers like cyclodextirins which are commonly used to minimize the ulcerogenic effect of various NSAIDs on the stomach besides its key role as solubility enhancers for poorly soluble lipophilic drugs [25-27].
Solid dispersion can be defined as a type of solid state material where molecular dispersion of one or more pharmaceutically active drugs are carried by an inert carrier. Particles in the form of solid dispersion are found to have higher degree of porosity [28-32]. Solid dispersion technique has been reported to be highly successful in improving the solubility and bioavailability of poorly soluble drugs.
The above mentioned considerations led to the objective of this study, which aimed to prepare and evaluate solid dispersion systems containing aceclofenac and Eudragit L100, Eudragit S100, Eudragit RL100, PVP K90, and methyl cellulose 15cps.
Materials and Methods
Materials
Aceclofenac (GMBH, Germany) was a gift sample kindly supplied by EPICO pharmaceuticals industries, El-Asher, Egypt, Eudragit L100, Eudragit S 100 and Eudragit RL100 were purchased from Sigma- Aldrich, St. Louis, Mo, USA. PVP k90 and MC cp15 were obtained from RÖhm Pharma GMBH, Darmstadt (Germany). All other reagents and chemicals were analytical grades and were used as received.
Aceclofenac (GMBH, Germany) was a gift sample kindly supplied by EPICO pharmaceuticals industries, El-Asher, Egypt, Eudragit L100, Eudragit S 100 and Eudragit RL100 were purchased from Sigma- Aldrich, St. Louis, Mo, USA. PVP k90 and MC cp15 were obtained from RÖhm Pharma GMBH, Darmstadt (Germany). All other reagents and chemicals were analytical grades and were used as received.
Methods
Preparation of solid dispersion systems
Solid dispersions using solvent evaporation technique were employed to coat aceclofenac with Eudragit L100, Eudragit S 100 , Eudragit RL100, PVP k90 and Methyl cellulose cp15 at different drug to polymer weight ratios of 1:1, 1:2 and 1:3. All formulations were prepared by dissolving the appropriate amount of the polymer in a mixture of isopropanol and acetone (1:1 v/v), with continuous stirring using magnetic stirrer (Stuart, Germany). An amount of aceclofenac equivalent to 100 mg was dissolved in a minimal amount of the solvent mixture at 40°C. The polymer solution was added gradually to the drug solution over a period of five minutes with continuous stirring. Organic solvents were allowed to evaporate over a period of 24 hrs under stirring conditions (150 rpm) at room temperature till a dry film was obtained. The resultant film was left in an oven for an additional 2 hrs at 40°C to ensure a full removal of the organic solvents from the samples [33-35]. The dried film was then inspected microscopically to observe any grittiness or drug precipitation. The dry film formed was granulated through a sieve (450 μm) (Fritsch Gmbh, Germany) in order to obtain drug granules with a homogenous particle size which eventually were stored at room temperature in dark tight containers in a desiccator over anhydrous calcium chloride [36] till used. Table I represents the composition of the prepared formulations
Preparation of solid dispersion systems
Solid dispersions using solvent evaporation technique were employed to coat aceclofenac with Eudragit L100, Eudragit S 100 , Eudragit RL100, PVP k90 and Methyl cellulose cp15 at different drug to polymer weight ratios of 1:1, 1:2 and 1:3. All formulations were prepared by dissolving the appropriate amount of the polymer in a mixture of isopropanol and acetone (1:1 v/v), with continuous stirring using magnetic stirrer (Stuart, Germany). An amount of aceclofenac equivalent to 100 mg was dissolved in a minimal amount of the solvent mixture at 40°C. The polymer solution was added gradually to the drug solution over a period of five minutes with continuous stirring. Organic solvents were allowed to evaporate over a period of 24 hrs under stirring conditions (150 rpm) at room temperature till a dry film was obtained. The resultant film was left in an oven for an additional 2 hrs at 40°C to ensure a full removal of the organic solvents from the samples [33-35]. The dried film was then inspected microscopically to observe any grittiness or drug precipitation. The dry film formed was granulated through a sieve (450 μm) (Fritsch Gmbh, Germany) in order to obtain drug granules with a homogenous particle size which eventually were stored at room temperature in dark tight containers in a desiccator over anhydrous calcium chloride [36] till used. Table I represents the composition of the prepared formulations
| Formula | Polymers used in solid dispersion | Drug: polymer ratio |
| F1 | Eud L 100 | 1:1 |
| F2 | 1:2 | |
| F3 | 1:3 | |
| F4 | Eud S 100 | 1:1 |
| F5 | 1:2 | |
| F6 | 1:3 | |
| F7 | Eud RL 100 | 1:1 |
| F8 | 1:2 | |
| F9 | 1:3 | |
| F10 | PVP K90 | 1:1 |
| F11 | 1:2 | |
| F12 | 1:3 | |
| F13 | MC 15 cps | 1:1 |
| F14 | 1:2 | |
| F15 | 1:3 |
Table 1: Composition of the prepared formulations.
Characterization of the prepared systems
A. Fourier transform infrared spectroscopy (FT-IR):
FT-IR spectrum studies help to confirm the identity of the drug and also to detect the interaction of the drug with the polymers (if any). FT-IR spectral measurements for free aceclofenac, polymers, and solid dispersions were carried out in order to find out any incompatibility between the drug and polymers. The process was carried out using FTIR analyzer (Perkin Elmer model, USA) adopting the KBr disk technique. All samples were grinded and mixed thoroughly with potassium bromide at a ratio of 1:100 (sample/KBr) followed by compressing the powders under pressure of 5 tons for 5 min using hydraulic press to form the KBr disk. Scans were obtained from 4000 to 450 cm-1 at a resolution of 2 cm-1 [37, 38].
A. Fourier transform infrared spectroscopy (FT-IR):
FT-IR spectrum studies help to confirm the identity of the drug and also to detect the interaction of the drug with the polymers (if any). FT-IR spectral measurements for free aceclofenac, polymers, and solid dispersions were carried out in order to find out any incompatibility between the drug and polymers. The process was carried out using FTIR analyzer (Perkin Elmer model, USA) adopting the KBr disk technique. All samples were grinded and mixed thoroughly with potassium bromide at a ratio of 1:100 (sample/KBr) followed by compressing the powders under pressure of 5 tons for 5 min using hydraulic press to form the KBr disk. Scans were obtained from 4000 to 450 cm-1 at a resolution of 2 cm-1 [37, 38].
B. Differential scanning calorimetry (DSC):
The melting point and heat of fusion of unprocessed/raw and processed drug was determined by using calorimeter (DSC-50, Shimadzu, Kyot, Japan). Approximately 2mg of powder sample was placed in a hermetically sealed aluminum pan (50 μl) with a pinhole at argon purge of 20 mL/min. The temperature difference between the sample and the reference is represented graphically in relation to the differential heat flow. The scanning rate of 20°C/min, from 40°C to 200°C was used in presence of argon [39].
The melting point and heat of fusion of unprocessed/raw and processed drug was determined by using calorimeter (DSC-50, Shimadzu, Kyot, Japan). Approximately 2mg of powder sample was placed in a hermetically sealed aluminum pan (50 μl) with a pinhole at argon purge of 20 mL/min. The temperature difference between the sample and the reference is represented graphically in relation to the differential heat flow. The scanning rate of 20°C/min, from 40°C to 200°C was used in presence of argon [39].
C. Powder X- ray Diffraction Analysis (XRD)
For crystallinity of the drug, samples were evaluated using X-ray powder diffraction, this was done by X-ray diffractometer (Bruker AXS model D8 Advance, Germany) under the following conditions: target Cu; filter Ni; voltage 40kv; current 40mA; receiving slit 0.2 inches. The data were collected in the continuous scan mode using a step size of 0.01° at 20/s. the scanning range was 5-50° at a wave length of 1.54°A. Samples used for XRD analysis were exactly the same as those used for DSC analysis [39].
For crystallinity of the drug, samples were evaluated using X-ray powder diffraction, this was done by X-ray diffractometer (Bruker AXS model D8 Advance, Germany) under the following conditions: target Cu; filter Ni; voltage 40kv; current 40mA; receiving slit 0.2 inches. The data were collected in the continuous scan mode using a step size of 0.01° at 20/s. the scanning range was 5-50° at a wave length of 1.54°A. Samples used for XRD analysis were exactly the same as those used for DSC analysis [39].
Drug content determination
The entrapment efficiency of the drug in the proposed drug delivery systems was determined after separation of the drug from solid dispersion. The entrapment efficiency (EE) was calculated using The entrapment efficiency of the drug in the proposed drug delivery systems was determined after separation of the drug from solid dispersion. The entrapment efficiency (EE) was calculated using the equation:
PEE = Total drug added-amount of the drug X 100
Total Drug added
The entrapment efficiency of the drug in the proposed drug delivery systems was determined after separation of the drug from solid dispersion. The entrapment efficiency (EE) was calculated using The entrapment efficiency of the drug in the proposed drug delivery systems was determined after separation of the drug from solid dispersion. The entrapment efficiency (EE) was calculated using the equation:
PEE = Total drug added-amount of the drug X 100
Total Drug added
Drug content in the proposed drug delivery systems was determined by crushing a known amount of solid dispersion in a mortar with a pestle before soaking in 100 ml phosphate buffer (pH 7.4) with continuous stirring using overheat stirrer (Stuart, Germany) for 90 min this provided complete swelling and bursting of the matrices. The resultant dispersions were filtered through 0.45 µm Millipore filter and the concentration of the drug in the solution was determined spectrophotometric ally after appropriate dilution using phosphate buffer (pH 7.4) as a blank [40]. The drug content was calculated as the percentage drug load as given by the formula [41-43].
% drug load = WD X 100
WB
% drug load = WD X 100
WB
Where WD is the amount of the drug loaded in the solid dispersion and WB is the weight taken of the proposed matrices. Drug content as performed in triplicate for each sample and the results were reported as a mean ± SD.
In- vitro drug release studies
The dissolution of the drug from the proposed drug delivery systems was carried out according to a united state pharmacopeia (USP) XXIV 8- station dissolution rate apparatus (Erweka type DT. Germany). One hundred mg of aceclofenac or its equivalent from the solid dispersion was packed in 5 size transparent hard gelatin capsules and the drug release was studied in 0.1N HCl (pH 1) and in phosphate buffer of pH value of 7.4. Samples of 5 ml were withdrawn at predetermined time intervals, filtered through Millipore filter (0.45 µm) and then assessed spectrophotometrically at a wavelength of 275 nm with a UV spectrophotometer (SHIMADZU, UV- 160A, Japan). Sample volume used for analysis was replaced by equal volumes of fresh dissolution medium preheated at 37°C to maintain the sink conditions.
The dissolution of the drug from the proposed drug delivery systems was carried out according to a united state pharmacopeia (USP) XXIV 8- station dissolution rate apparatus (Erweka type DT. Germany). One hundred mg of aceclofenac or its equivalent from the solid dispersion was packed in 5 size transparent hard gelatin capsules and the drug release was studied in 0.1N HCl (pH 1) and in phosphate buffer of pH value of 7.4. Samples of 5 ml were withdrawn at predetermined time intervals, filtered through Millipore filter (0.45 µm) and then assessed spectrophotometrically at a wavelength of 275 nm with a UV spectrophotometer (SHIMADZU, UV- 160A, Japan). Sample volume used for analysis was replaced by equal volumes of fresh dissolution medium preheated at 37°C to maintain the sink conditions.
In-vivo ulcerogenicity studies
Experimental animals
Male Wistar rats, weighing 180-200gm, were obtained from National researches center (Cairo, Egypt). Rats were maintained at 22±1°C on a 12 hrs light-dark cycle allowed rat chow and water ad libitum.
Experimental animals
Male Wistar rats, weighing 180-200gm, were obtained from National researches center (Cairo, Egypt). Rats were maintained at 22±1°C on a 12 hrs light-dark cycle allowed rat chow and water ad libitum.
The allocation of animals to all groups was randomized. In-vivo experimental protocols had the approval of the institutional animal ethics committee (IAEC) (IAEC/PROPOSAL/DB-4/2010).
Before the start of the experiments, rats were housed individually in wire mesh cages to avoid coprophagy under controlled environmental conditions. Food was withdrawn for 36 hrs but water was allowed ad libitum [44]. The absence of ulcers in some of the treated groups has revealed that the pre- fasting conditions alone doesn't induce ulcer. Table (2) shows the experimental design and animal groups.
| Group Number | Treatment |
| (Control group) | Rats were orally administered (p.o.) 1 ml distilled water |
| Aceclofenac (20mg/kg) | Rats were (p.o.) 1ml aceclofenac solution |
| Aceclofenac - Eudragit S 100 solid dispersion | Rats were (p.o.) 1ml of aceclofenac - Eudragit S 100 solid dispersion |
Table 2: Experimental design and animal groups.
As described in the previous studies [45-47] on the morning of the experiments each fasted rat was orally administered 1 ml suspension of the assigned drug by oral gavage in a dose equivalent to 20 mg per kg of aceclofenac or different aceclofenac solid dispersion systems [48]. Magnetic stirring was utilized to obtain a well-dispersed suspension of each drug and solid dispesion. Six hours later, each animal was anaesthetized with ether, and the abdomen was opened. This time interval for drug administration was subjected to the time table required for induction of peptic ulcer according to Saheed et al., [49] who used Indomethacin as an example of NSAIDs which induced peptic ulcer. Each stomach was excised, dissected along the greater curvature and contents were emptied by gently rinsing with isotonic saline solution.
Histopathological examination of stomach sections
For histopathological examination, the stomach was surgically extirpated from each group and opened through vertical incision along the greater curvature and photographs were taken of the inside surface of the stomach. The stomach tissues were then washed in 0.9% saline and a portion of it was kept in 10% buffered formalin solution for histopathological studies [50, 51]. The sections were then stained with hematoxylin and eosin. The tissue sections were examined under an Olympus BX51 (Olympus Corporation, Tokyo, Japan) formulations and images were captured with a digital camera attached to the formula [52].
For histopathological examination, the stomach was surgically extirpated from each group and opened through vertical incision along the greater curvature and photographs were taken of the inside surface of the stomach. The stomach tissues were then washed in 0.9% saline and a portion of it was kept in 10% buffered formalin solution for histopathological studies [50, 51]. The sections were then stained with hematoxylin and eosin. The tissue sections were examined under an Olympus BX51 (Olympus Corporation, Tokyo, Japan) formulations and images were captured with a digital camera attached to the formula [52].
Statistical analysis
One way ANOVA test followed by Tukey posttest was used for comparisons between the treatment and control groups. Data were presented as Mean ± SD. The (P values < 0.05) was considered as significance level during this study.
One way ANOVA test followed by Tukey posttest was used for comparisons between the treatment and control groups. Data were presented as Mean ± SD. The (P values < 0.05) was considered as significance level during this study.
Results and Discussion
Fourier transform infrared spectroscopy
Figure 2: Shows IR spectrum of the drug as well as in the proposed formulations.
Figure 2: Shows IR spectrum of the drug as well as in the proposed formulations.

Figure 2: IR spectra of aceclofenac, polymers used, solid dispersions formulations.
It is obvious that the spectrum of the free drug showed characteristic bands at 3318 cm-1 (N-H stretching), 2970-1 cm and 2930 cm-1 (O-H stretching), 1719 cm-1 (C=O stretching), 1585 cm-1 (aromatic C=C stretching), 1506 cm-1 (in plane bending for N-H), 965 cm-1 (O-H out plane bending) and 750 cm-1 (N-H out plane bending). Eudragit S 100 produced a characteristic absorption band at 3481 cm-1 (O-H stretching), 1728 cm-1 (C=O stretching). Eudragit L100 the spectrum characterized by the absorption at 3473 cm-1 (O-H stretching), 1738 cm-1 (C=O stretching). Eudragit RL 100 showed a characteristic absorption band at 3439 cm-1 (O=H stretching), 1733 cm-1 (C=O stretching). In case of PVP K90 the spectrum was characterized by absorption band at 3444 cm-1 (O- H stretching), 1659 cm-1 (C=O stretching).
While the FTIR spectrum of MC showed absorption band at 3452 cm-1 (O-H stretching), 1639 cm-1 (C=O stretching).
The FTIR spectrum of the solid dispersion of aceclofenac with Eudragit S100 and Eudragit L100 show the main absorption band of aceclofenac but shifted to upper frequencies. This means no interaction between the drug and polymers.
In case of Eudragit RL 100, MC and PVP K90 revealed distinct peak of aceclofenac but the band corresponding to the -NH group overlapped with that of the -OH group of the [53] and carbonyl group in case of PVP K90 indicate no interaction between the drug and polymers used.
Differential Scanning Calorimetry
Figure 3 shows DSC thermograms of aceclofenac solid dispersion systems with different polymers in comparison to the free drug.
Figure 3 shows DSC thermograms of aceclofenac solid dispersion systems with different polymers in comparison to the free drug.
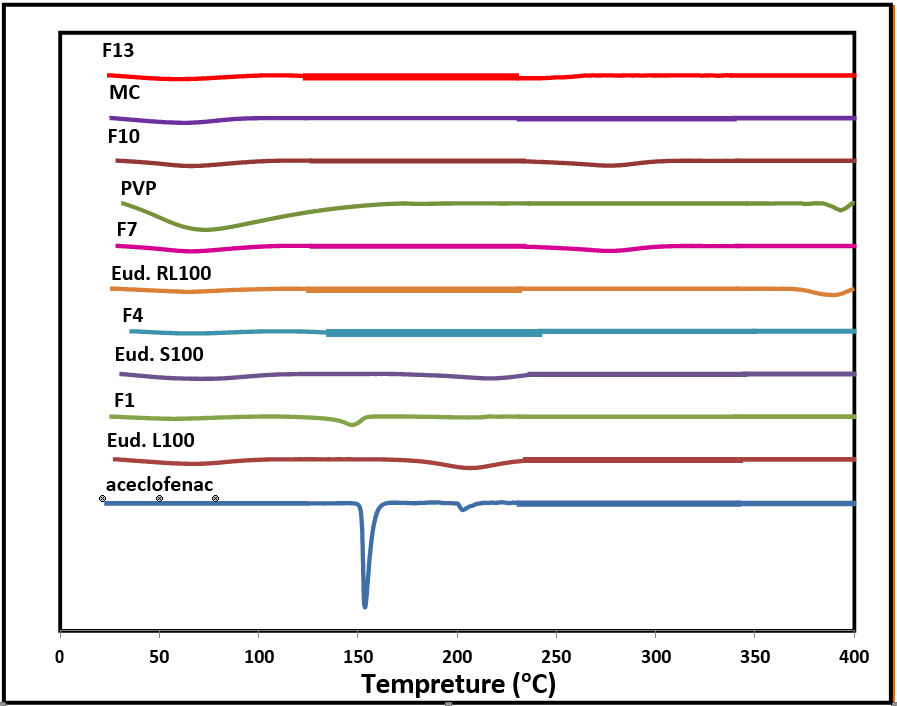
Figure 3: DSC thermograms of aceclofenac, polymers used, solid dispersions formulations.
DSC thermograms confirmed no occurrence of any prominent interaction between the drug and the polymers. Free drug showed a characteristic endothermic peak with a Tm recorded at 153.46°C revealing that the unprocessed free aceclofenac present in the crystalline state which is corresponding to the published data [54]. The recorded thermogram of Eudragit L100 produced a broad endothermic peak with an onset of 29.15°C and ends at 97.25°C this thermal curve was attributed to trace moisture. Eudragit S100 thermogram appears to be a broad peak with an onset of 30.95°C and ends at 107.04°C belongs to adsorbed moisture which is in agreement with published data [55]. The thermal behavior of Eudragit RL100 presents a broad peak at 64.67°C [56]. For a PVP, a strong broad peak starts at 33.29°C and ends at 144.85°C due to adsorbed moisture [53] the thermal curve of methyl cellulose is characterized by a weak broad peak starts at 27.44°C and ends at 91.32°C which can be attributed to bounded moisture [57]. Preparation of aceclofenac by solid dispersion gives drug crystals with thermograms depending on the polymer used. Formulations of aceclofenac with Eudragit L100, Eudragit S100 and Eudragit RL100 present weak endothermic peaks at 147.03°C, 149.67°C and 121.69°C respectively. This behavior reflects the change in the crystalline state of drug to partially amorphous or drug distributed in polymer matrix at molecular level [58]. In case of aceclofenac with PVP which is characterized by disappearance of drug peak this can be explained as possible transition from crystalline to amorphous state. Finally the recorded thermogram of drug with methyl cellulose produced a weak endothermic peak at 139.96°C also suggests a change in crystalline form of the drug or it is dissolved in polymer matrix [59].
Powder X- ray Diffraction Analysis
Figure 4 shows the diffractograms of the free aceclofenac, polymers and solid dispersion preparations.
Figure 4 shows the diffractograms of the free aceclofenac, polymers and solid dispersion preparations.
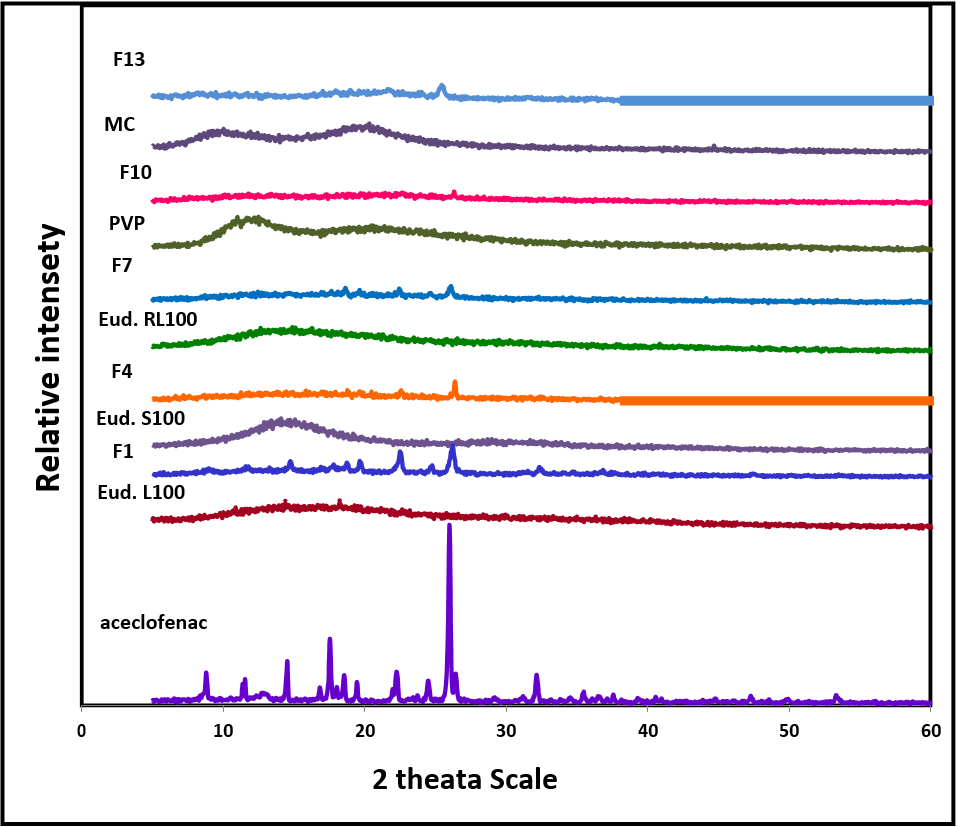
Figure 4: X-ray diffractogram of aceclofenac, polymers used, solid dispersions formulations.
The diffraction pattern of free drug shows its crystalline nature by numerous clear peaks at a diffraction angle (2ø) of 8.89°, 11.58°, 12.96°, 14.61°, 16.94°, 17.65°, 18.11°, 19.55°, 22.37°, 24.60°, 26.09°, 26.54°, 32.27°, 35.59°, 36.68°, 47.41° and 53.49° this indicate the crystalline nature of aceclofenac. The diffractogram of Eudragit S100, EudragitL100, Eudragit RL100, PVPK90 and MC show amorphous structure. The solid dispersion of aceclofenac with these polymers shows weak peaks at 22.52° and 26.20° in Eudragit L 100, 26.42° in Eudragit S 100, 22.51° and 26.12° in Eudragit RL 100, 25.44° in MC. The diffraction pattern of solid dispersion with PVP K90 indicates weakest peak at 22.65°.
This can be attributed to reduction in particle size of aceclofenac after solvent evaporation; the drug is dispersed in the polymer matrix at a molecular level giving possibility of partial amorphisization.
Drug content determination:
Aceclofenac content in different formulations is presented in Table 3
Aceclofenac content in different formulations is presented in Table 3
| Formula | % Aceclofenac Content* |
| F1 | 97 ± 0.98 |
| F2 | 98 ± 0.74 |
| F3 | 98 ± 1.88 |
| F4 | 99 ± 0.72 |
| F5 | 97 ± 1.93 |
| F6 | 100 ± 2.14 |
| F7 | 99 ± 3.23 |
| F8 | 98 ± 2.36 |
| F9 | 99 ± 1.57 |
| F10 | 99 ± 1.23 |
| F11 | 99 ± 0.64 |
| F12 | 98 ± 0.13 |
| F13 | 98 ± 1.62 |
| F14 | 98 ± 0.87 |
| F15 | 100± 2.27 |
* (Mean ± SD, n=3)
Table 3: Aceclofenac content in different solid dispersion systems.
Table 3: Aceclofenac content in different solid dispersion systems.
From the Table it is obvious that drug content in the prepared formulations varied from 97.98 to 100. It is also evident from the table that drug to polymer as well as the method of preparation didn’t play any role on drug content in the proposed formulations. The obtained results are in parallel with those obtained by Swetha et al, [60] who prepared micro sponges containing etodolac with different types of polymers including Eudragit and ethyl cellulose and proved that the ratio of the polymer in the delivery system has no effect on the percentage entrapment efficiency. Trivedi et al [61] reported that by increasing the polymer ratio in certain formulations from 1:1 to 1:5 was followed by increasing drug entrapment efficiency.
In-vitro drug release studies:
Release characteristics of the drug from solid dispersion systems are presented in figures 5 and 6
Release characteristics of the drug from solid dispersion systems are presented in figures 5 and 6
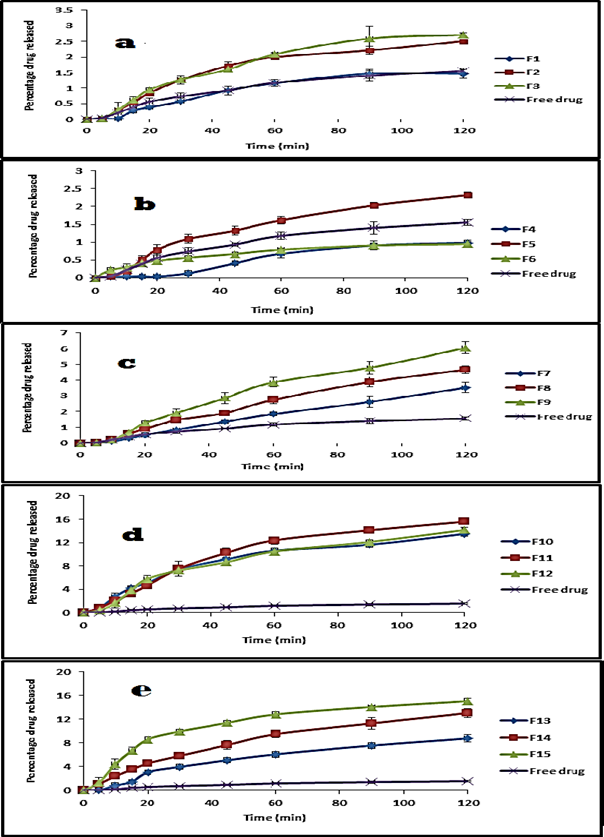
Figure 5: Release profile of aceclofenac as a free drug and from its solid dispersion systems using Eud. L100 (a), Eud. S100 (b), Eud. RL100 (c), PVP k90 (d), MC 15cp (e) at pH 1 (0.1N HCl).
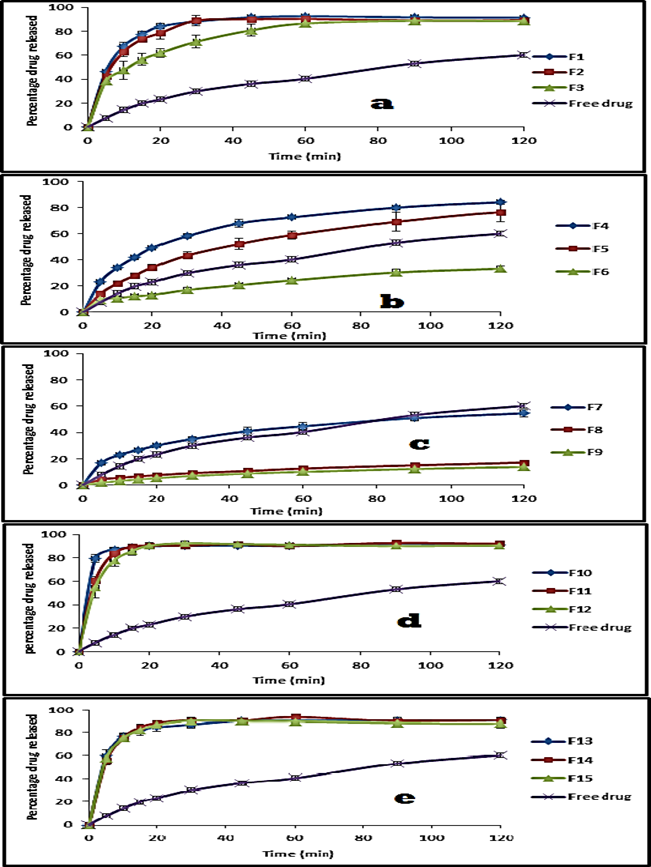
Figure 6: Release profile of aceclofenac as a free drug and from its solid dispersion systems using Eud. L100 (a), Eud. S100 (b), Eud. RL100 (c), PVP k90 (d), MC 15cp (e) at pH 7.4 (phosphate buffer).
From the above figures, it is obvious that the percentage dissolved after 20 min of the free drug was 0.56 and 23.07%. At time 120 min it was 1.65 and 81.20% at pH 1 and 7.4 respectively. Formulation of solid dispersion of aceclofenac with Eud L100 resulted in a non-significant decrease in the dissolution rate compared to the free drug, after 20 minute was 0.39±0.02 and after 120 min was 1.46±0.14% at pH 1 and at drug: polymer ratio 1:1, while showed significant increase in the dissolution rate of compared to the free drug at 20 min 84.02±2.26% and at 120 min 91.45±0.22% at pH 7.4 (p value <0.05). So the amount of the drug dissolved in the 20 min was increased gradually with increasing polymer concentration to reach 0.85±0.05% and 0.95±0.05%, at 120 min, 2.51±0.06% and 2.71±0.05% at pH 1, at drug: polymer ratio 1:2 and 1:3 respectively (p value <0.05). At pH 7.4, the amount of drug dissolved at 20 min was decreased gradually with increase polymer concentration to reach 78.53±5.19% and 61.95±3.66%, at 120 min 89.3±2.09% and 88.45±0.49% at drug: polymer ratio 1:2 and 1:3 respectively(p value <0.05). So the amount of the drug dissolved in 120 min at pH 7.4 decreased due to the enteric effect of Eud L100, this can be attributed to ionization of the carboxylic group in aqueous media at pH 5.5 and above, rendering the polymer resistant to acidic media [62]. Such polymers retain the drug when in contact with gastric environment and release the drug rapidly in intestinal environment.
As for Eud L100, preparation of solid dispersion of aceclofenac with Eud S100 resulted in significant decrease in the dissolution rate of the product at 20 min was 0.03% and at 120 min was 0.98±0.08% at pH 1, while resulted in significant increase in dissolution rate at 20 min which was 49.35±0.65% and at 120 min was 84.19±1.76% at pH 7.4 compared to the free drug. So the amount of the drug dissolved in the 20 min was increased gradually with increasing polymer concentration to reach 0.79±0.15% and 0.48±0.03%, at 120 min reach 2.32±0.07% and 0.94±0.07% at pH 1, at 1:2 and 1:3 respectively(p value <0.05). At pH 7.4, the amount of drug dissolved at 20 min was decreased gradually with increase polymer concentration to reach 34.3±2.04% and 13.24±0.86%, at 120 min reach 76.3±7.11% and 33.36±1.91% at 1:2 and 1:3 respectively (p value <0.05). This can be attributed to physical change in the crystalline structure of the drug.
For Eud. RL100, formulation of solid dispersion with aceclofenac showed non-significant decrease in the dissolution rate of the product at 20 min 0.52±0.02%, but show significant increase in the dissolution rate at 120 min 3.52±0.32% at pH 1, while resulted in non-significant increase 29.9±0.96%at 20 min and no significant decrease 54.5±2.72% at 120 mint compared to the free powder at pH value 7.4 in a drug: polymer ratio of 1:1. At pH 1 increase the drug: polymer ratio showed increase the dissolution rate of product at 20 min and 120 min in both ratios 1:2 and 1:3, on the other hand at pH 7.4, increase the concentration of the polymer resulted in decreasing the amount of drug released at 20 min and 120 min in both ratios drug: polymer ratio with 1:2 and 1:3 as showed in figures 5 and 6. This can be attributed to its time dependent property, not pH dependent like Eud L100 and Eud S100.
In PVP K90, the product showed significant increase in the dissolution rate compared with free drug at 20min 5.08±0.41% and 13.5±0.44% at 120 min at pH 1, also showed significant increase in the dissolution rate at 20 min 90.03±0.6% and at 120 min 91.11±1.05% compared to free drug at pH 7.4 in drug: polymer ratio of 1:1(p value <0.05). Increase the polymer concentration resulted in significant increase in the dissolution rate at 20 min and 120 min in both gastric and intestinal media at drug: polymer ratio of 1:2 and 1:3 drugs to polymer ratio as show in in figures 5 and 6. This can be attributed to increase the wheat ability and decrease hydrophobicity. The presence of hydrophilic polymer may prevent aggregation of fine particles exposing a higher surface area of drug to the dissolution media [63].
Finally, in case of methylcellulose (MC) the product resulted in significant increase in the dissolution rate 3.07±0.33% at 20 min and 8.79±0.63% at 120 min being compared with free drug at pH 1, The dissolution rate of product showed significant increase at 20 min 84.7±3.62% and 90.43±2.23% at 120 min at pH 7.4 at 1:1 drug: polymer ratio (p value <0.05).
Eud. S 100 at a drug: polymer ratio of 1:1 was selected as the optimal solid dispersion using enteric polymer to conduct further in vitro and in vivo evaluation, since it was the lowest ratio which achieved significant reduction in drug release at both fasted and fed state of the stomach while performing higher dissolution at intestinal pH and consequently higher drug bioavailability.
Histopathological results
The pattern of the mucosal specimens was studied histopathologically by examining the histology of the treated and control samples. Effect of aceclofenac, its solid dispersion formula on stomach tissue histopathology is presented in Figure 7
The pattern of the mucosal specimens was studied histopathologically by examining the histology of the treated and control samples. Effect of aceclofenac, its solid dispersion formula on stomach tissue histopathology is presented in Figure 7

Figure 7: Representative image showing histopathological observations in rat gastric tissues after administration of aceclofenac and its solid dispersions, (a) control group, (b) free drug and (c) drug- Eud S100 solid dispersion formula.
Histopathological examination of Hx & E stained stomach sections of rats administered distilled water (control group), showed that all the four animals appeared with full thickness of gastric wall (normal mucosa, musculosa and serosa), normal gastric mucosa consisting of glands lined with mucin secreting cells (Figure 7-a). In rats administered aceclofenac (20mg/kg), histopathological examination showed that all the four animals, evidenced loss of gastric mucosa and replaced by granulation tissue, foci exhibiting chronic inflammation and intestinal metaplasia of gastric glands denoting areas of gastritis and parietal cells hyperplasia (Figure 7-b). In rats administered aceclofenac- Eudragit S100 solid dispersion, histopathological examination detected that stomach tissue of animals manifested focal partial loss of gastric mucosa, (Figure 7-c).
The integrity of the gastric mucosa depends on the balance between aggressive (HCl, pepsine) and protective factors (mucus and HCO3-secretion, prostaglandins, mucosal blood flow, nitric oxide) [64].
The treatment is effective depending not only on the blockade of acid secretion, but also on the increased production of factors responsible for protecting the gastric mucosa, thus avoiding damage to the epithelium [65].
Inhibition of prostaglandin synthesis is well recognized as the central mechanism by which gastrointestinal injury occurs [66]. This is a result of inhibition of cyclooxygenase enzyme which converts unsaturated fatty acids (which are released during cell injury) such as arachidonic acid to prostaglandins. In the stomach, prostaglandin synthesis is protective as a result of enhanced mucosal blood flow and stimulation of mucous and bicarbonate secretion [67].
In contrast, in arthritis, prostaglandins mediate pain and some components of inflammation. Recognition of two isoforms of cyclooxygenase, with COX1-predominating in the stomach and an inducible COX-2 expressed at sites of inflammation offer the prospect of separating the beneficial effects of inhibiting prostaglandin synthesis in joints from the harmful effects of inhibiting it in the stomach [68].
The primary objective of the present investigation was to determine whether the enteric-polymers provide protection against aceclofenac -induced damage to gastric mucosa. Results showed that the enteric-polymers used in this study are capable of providing protection to the gastric mucosa against aceclofenac -induced gastric injury. In most of our experiments, the aceclofenac -induced gastric ulceration was maximally protected by coating with enteric-polymers at the dose of 20mg/kg (fed orally).
Conclusion
The obtained results of solid dispersion systems indicated that solid dispersion of aceclofenac with EudS100 as an enteric carrier and its slow diffusion into the gastric lumen as confirmed by in vitro dissolution data could alleviate the problem of gastric ulceration by minimizing its direct exposure to the ulcer-prone area of the stomach. Solid dispersion characterization using FT-IR, DSC and XRD revealed that no significant changes occurred for aceclofenac. Solid dispersion of aceclofenac using EudS100 at drug to polymer ratio of 1:1significantly reduced gastric irritations and gastric ulcers compared to the free drug.
References
- Mutalik S, Anju P, Manoj K, Usha A N. (2008). "Enhancement of dissolution rate and bioavailability of aceclofenac: A chitosan-based solvent change approach" Int. J. Pharm, 350 (1): 279-290.
- Naglakshmi S, Shanmuganathan S, Sandhya K, Anbarasan B. (2018). "Design, development and characterization of nano structured lipid carrier for topical delivery of aceclofenac" Ind. J. Pharm. Edu. Res., 52(4): 581- 586.
- Wolfe M, Lichtenstein D, Singh G., (1999). "Gastrointestinal toxicity of non-steroidal anti- inflammatory drugs." N Engl J Med, 340 (24): 1888-99.
- Wolfe M, Andrew H, Soll, M, (1988). "The physiology of gastric acid secretion" N Engl J Med, 31: 1707-15.
- Allen A, Flemström G, Garner A, Kivilaakso E. (1993). "Gastro-duodenal mucosal protection." Physiol Rev, 73: 823-857.
- Schoen R, Vender R. (1989). "Mechanisms of non-steroidal anti-inflammatory drug-induced gastric damage." Am. J. Med, 86: 449-58.
- Somasundaram S, Hayllar H, Rafi S, Wrigglesworth J, Macpherson A, Bjarnason I. (1995). "The biochemical basis of non-steroidal anti-inflammatory drug-induced damage to the gastrointestinal tract: a review and a hypothesis." Scand J Gastroenterol, 30: 289-299.
- Darling R, Romero J, Dial E, Akunda J, Langenbach R, Lichtenberger L. (2004). "The effect of aspirin on gastric mucosal integrity, surface hydrophobicity, prostaglandin metabolism in cyclooxygenase knockout mice." Gastroenterology, 127: 94-104.
- Lichtenberger L, Zhou Y, Dial E, Raphael R. (2006). "NSAID injury to the gastrointestinal tract: evidence that NSAIDs interact with phospholipids to weaken the hydrophobic surface barrier and induce the formation of unstable pores in membranes." J. Pharm. Pharmacol, 58: 1421-1428.
- Lanas A. (2010). "A review of the gastrointestinal safety data-a gastroenterologist’s perspective. Rheumatology" (Oxford), 49(2): ii3–ii10.
- Silverstein F, Faich G, Goldstein J. (2000). "Gastrointestinal toxicity with celecoxib vs non-steroidal anti-inflammatory drugs for osteoarthritis and rheumatoid arthritis: the CLASS study: A randomized controlled trial. Celecoxib Long-term Arthritis Safety Study." JAMA, 284: 1247-55.
- Yong C, Oh Y, Lee K, Park S, Park Y, Gil Y. (2005). "Trials of clear aceclofenac-loaded soft capsules with accelerated oral absorption in human subjects." Int. J. Pharm, 302: 78-83.
- Joshi V, Sawant M., (2006). "Study on dissolution rate enhancement of poorly water soluble drug: contributions of solubility enhancement and relatively low micelle diffusivity." J. Disp Sci Technol, 27(8): 1141-50.
- Dahiya S, Pathak K., (2006). "Physicochemical characterization and dissolution enhancement of aceclofenac-hydroxypropyl betacyclodextrin binary systems." PDA J. Pharm. Sci. Technol, 60 (6): 378-88.
- Desai S, Nadkarni S, Wald R, Debrincat G., (2007). "Dual release compositions of a cyclooxygenase-2 inhibitor." US Patent, 7220434.
- Muatlik S, Usha A, Reddy M, Ranjith A, Pandey S., (2007). "Improved bioavailability of aceclofenac from spherical agglomerates: development, in vitro and preclinical studies." Pak J. Pharm. Sci. 20(3): 218-26.
- Yesmin F, Talukder M, Islam M, Laila S, Haque T. (2008). "Evaluation of aceclofenac loaded agarose beads prepared by ionotropic gelation method." S J Pharm Sci, 1(12):10-17.
- Kilor V, Sapkal N, Awari J, Shewale B. (2010). "Development and characterization of enteric-coated immediate-release pellets of aceclofenac by extrusion/spheronization Technique Using κ- carrageenan as a pelletizing agent." AAPS Pharm Sci Tech, 11: 336-343.
- Margret Chandira R, Venkataeswarlu B, Kumudhavalli M, Bhowmik D, Jayakar B. (2008). "Formulation and evaluation of the fast dissolving tablets of aceclofenac." The Pharma Review, 164-67.
- Lin SY, Kawashima Y. (1987). Drug release from tablets containing cellulose acetate phthalate as an additive or enteric-coating material. Pharm Res, 4: 70-74.
- Venkatesan P, Manavalan R and Valliappan K. (2009). "Microencapsulation: A vital technique in novel drug delivery system." J. Pharm. Sci. Res., 1(4): 26-35.
- Bansode SS, Banarjee SK, Gaikwad DD, Jadhav SL, Thorat RM. (2010). "Microencapsulation: A review." Int. J. Pharm. Sci. Rev. Res., 1(2): 38-42.
- Hemanta KS, Siba PP, Babita S. (2010). "Preparation and in vitro evaluation of enteric controlled release pantoprazole loaded microcapsules using natural muco-adhesive Substance from Dilleniaindica L." Int. J.. Pharm. Tech. Res., 2(1): 542-551.
- Goudanavar P.S, Pagali R.S, Chandrashekhara S. (2010). "Design and characterization of tenoxicam microcapsules by ionotropic gelation technique." Int. J. Pharm. Bio Sci., 1(2): 1-10.
- Challa R, Ahuja A, Ali J, Khar R. (2005). "Cyclodextrins in Drug Delivery: An Updated Review." AAPS Pharm Sci. Tech, 6 (2): E329-E357.
- Loftsson T, Duchene D. (2007). "Cyclodextrins and their pharmaceutical applications." Int. J. Pharm, 329:1-11.
- Uekama K, Hirayama F, Tetsumi I. (1998). "Cyclodextrin drug carrier systems." Chem. Rev, 98: 2045-2076.
- Dharna Al, Singh N, Singh S, Arora S. (2013). "Solid dispersion a review on drug delivery system and solubility enhancement" Int. J. Pharm Sci. Res., 4 (6): 2049-2105.
- Dixit A K, Singh R P. (2012). "Solid dispersion a strategy for improving solubility of poorly soluble drugs." Int. J. Pharm. Sci., 3(2): 960-966.
- Singh S, Baghel R, Yadav L, (2011). "A review on solid dispersion" Int. J. Pharm. Life Sci., 2(7): 1078-1095.
- Srinarong P, Waard H, Frijlink H, Hinrichs W. (2011). "Improved dissolution behavior of lipophilic drugs by solid dispersions: the production process as starting point for formulation considerations.", Expert Opinion on Drug Delivery, 8: 1121-1140.
- Arunachalam A, Karthikeyan M, Konam K, Pottabathula P, S.Sethuraman, Ashutoshkumar S, (2010). "solid dispersion a review" Curr. Pharm. Res., 1: 1-10.
- Sera juddin, A., (1999). "Solid dispersion technique." J. Pharm. Sci, 88 (10): 891- 900.
- Rassu, G., Gavini, E., Spanda, G., Giunchedi, P., Morcedd, S., (2008). “Ketoprofen spray-dried microspheres based on Eudragit RS and RL study of the manufacturing parameters." Drug Dev. Ind. Pharm., 34(11):1178 -1187.
- Sudhamani, T., Reddy, K.N., Kumar, V.R., Revathi, R., Ganesan, V., (2010). "Preparation and evaluation of ethyl cellulose microspheres of Ibuprofen for sustained drug delivery." Int. J. Pharm. Res. Dev. 2 (8): 119 -125.
- Essa, E., Negm, M., Eldin, E. Z., & El-Maghraby, G. (2017). “Fast disintegrating tablets of amiodarone for oral administration." J. Applied Pharm. Sci., 7(1): 64-72.
- Dyer JR, "Absorption of common functional groups, Application of absorption spectroscopy of organic compounds." 7th. New Delhi, Prentice Hall of India, Pvt. Ltd, 1989; 32-37.
- Savita V, Piyush T, Subhash C. (2009). "Dextran -Etodolac conjugates: synthesis, in vitro and in vivo evaluation." Acta. Pol. Pharm. Drug. Res, 66 (2): 201-206.
- Pignatello, R., Ferro, M., De Guidi, G., Salemi, G., Vandelli, M.A., Guccione, S.m (2001). "Preparation, characterization and photosensitivity studies of solid dispersions of diflunisal and Eudragit RS 100 and RL 100." Int. J. Pharm. 218: 27-42.
- Singh, G., Pai, R.S., Devi, V.K. (2012). "Response surface methodology and process optimization of sustained release pellets using Taguchi orthogonal array design and central composite design." J. Adv. Pharm. Tech. Res. 3: 30-40.
- Viral, S., Hitesh, J., Jethva, K., Pramit, P., (2010). Micro sponge drug delivery. A Review. Int. J. Res. Pharm. Sci. 1(2): 212: 218.
- John, I.D., Harinath, N.M., (2008). Topical anti-Inflammatory gels of fluocinolone acetonide entrapped in Eudragit based micro-sponge delivery system. Res. J. Pharm. Tech. 1(4): 502-506.
- Ali, N., Mitra, J., Mohammed, Reza S., Siavoosh, D., (2005). "The effect of formulation types on the release of benzoyl peroxide from micro sponges." Iran. J. Pharm. Sci. 1(3): 131-142.
- El-shitany N, (2006). Mechanism of omeprazole induced gastric protection against ethanol-induced gastric injury in rats; Role of mucosal nitric oxide and apoptotic cell death. Proceeding of 1st international Egyptian-Jordanian conference on biotechnology and sustainable development: current status& future scenarios, Medical& Pharmaceutical, 2:183-193.
- Bhargava K, Gupta M, Tangri K. (1973). Mechanism of ulcerogenic activity of indomethacin and oxyphenbutazone. Eur. J. Pharmacol., 22(9): 95.
- Schmassmann A, Peskar B, Selter C. (1998). Effect of inhibition of prostaglandin endoperoxide synthase-2 in chronic gastro-intestinal ulcer models in rats. Br. J. Pharmacol., 123:795- 804.
- Brzozowski T, Konturek P, Konturek S. (2001). "Classic NSAIDs and selective cyclooxygenase (COX-1) and (COX-2) inhibitors in healing of chronic gastric ulcers." Microsc. Res. Tech., 1:343-53.
- Keumhan N, Beom S, Kwang-il Kwon S, Hwi-yeol Y, Eunyoung Kim T, Cheon Jeong W. (2015). "Absolute bioavailability and metabolism of aceclofenac in rats" Archives of Pharmacal Research, 38 (1): 68-72.
- Saheed S, Taofeeq G, Taofik S, Emmanuel A, Abdulhakeem S, Ismaila N, Abdulazeez B, (2015). ndomethacin-induced gastric ulceration in rats: Protective roles of Spondias mombin and Ficus exasperata Toxicology Reports 2: 261-267.
- Alsarra I, Ahmed M, Alanazi F, El-Tahir K, Alsheikh A, Neau S, (2010). Influence of Cyclodextrin Complexation with NSAIDs on NSAIDs/Cold Stress-Induced Gastric Ulceration in Rats. Int. J. Ned. Sci., 7: 232-239.
- Karanachi A, Reddy K, Degennaro D, Khan A. (1997). Comparative Evaluation of the severity of gastric ulceration by solid dispersions and coprecipitates of indomethacin. J. of Drug Targeting 4(5): 297-301.
- Zien E, El Rashidy M, Ghorab M, Gad S, Yassin H. (2015). "In vivo evaluation of ulcerogenic activity of ketorolac, its solid dispersion systems, as well as its microcapsules in rats. " J. Pham. Ph. Sci. 4(3): 23-37.
- El Maghraby, G. M., & Elsergany, R. N. (2014). "Fast disintegrating tablets of nisoldipine for intra-oral administration." Pharm. Dev. & Tech., 19(6): 641-650.
- Maulvi, F. A., Dalwadi, S. J., Thakkar, V. T., Soni, T. G., Gohel, M. C., & Gandhi, T. R. (2010). "Improvement of dissolution rate of aceclofenac by solid dispersion technique." Powder, 207: 47- 54
- Jablan, J., & Jug, M. (2015). "Development of Eudragit® S100 based pH-responsive microspheres of zaleplon by spray-drying: Tailoring the drug release properties." Powder Technology, 283: 334-343.
- Katara, R., & Majumdar, D. K., Eudragit RL 100-based nano-particulate system of aceclofenac for ocular delivery." Colloids and Surfaces B: Biointerfaces, 2013,103, 455- 462.
- Ozeki, T., Yuasa, H., & Okada, H. (2005). "Controlled Release of Drug via Methylcellulose- Carboxyvinyl polymer Inter-polymer Complex Solid Dispersion." AAPS Pharm. Sci. Tech. 6(2): 231–236.
- Sancin, P., Caputo, O., Cavallari, C., Passerini, N., Rodriguez, L., Cini, M., & Fini, A. (1999). "Effects of ultrasound-assisted compaction on Ketoprofen / Eudragit ® S100 Mixtures." Eur. J. Pharm. Sci., 7: 207–213.
- Essa, E. A., Elmarakby, A. O., Donia, A. M. A., & El Maghraby, G. M. (2017). "Controlled precipitation for enhanced dissolution rate of flurbiprofen: development of rapidly disintegrating tablets." Drug Dev. Ind. Pharm., 43(9): 1430-1439.
- Swetha, A., Gopal Rao, M., Venkata Ramana, K, Niyaz Basha Koti Reddy, V., (2011). "Preparation and evaluation of etodolac loaded Eudragit RS 100 microcapsules using quality by design approach." Int. J. Pharm. 1(2): 73 -80.
- Trivedi, P., Verma, A., Garud, N., (2008). "Preparation and characterization of aceclofenac microspheres." Asian J. Pharm. 2 (8): 119-125.
- Arun Raj, R. (2013). "Design and development of aceclofenac alginate beads for sustained release drug delivery." Int. J. Pharm Pharma. Sci., 5(1): 82-85.
- Betageri, G. V., & Makarla, K. R. (1995). “Enhancement of dissolution of glyburide by solid dispersion and lyophilization techniques." Int. J. Pharm., 126(1-2): 155-160.
- Lam E, Tai E, Koo M, Wong H. (2007). "Enhancement of gastric mucosal integrity by Lactobacillus rhamnosus." G.G. Life Sci., 80: 2128-2136.
- Moraes M, Kushima H, Moleiro C, Santos C. (2009). "Effects of limonene and essential oil from Citrus aurantium on gastric mucosa, role of prostaglandins and gastric mucus secretion." Chem. Bio. Int., 180: 499-505.
- Vane R. (1971). "Inhibition of prostaglandin synthesis as a mechanism of action for aspirin-like drugs." Nat New Biol., 231: 232- 235.
- Hawkey J. (2002). "Cyclooxygenase inhibition, between the devil and the deep blue sea." Gut 50: 25-30.
- Fu Y, Masferrer L, Seibert K, Raz A, Needleman P. (1990). "The induction and suppression of prostaglandin H2 synthase (cyclooxygenase) in human monocytes." J. Biol. Chem. 265 (16): 737-740.
Citation: Esmat E. Zein., et al. (2019). Effect of Certain Polymeric Resins as Well as Water Soluble Carriers on the Release of Aceclofenac and its Ulcerogenic Activity in Rats. Journal of Pharmacy and Drug Development 1(2).
Copyright: © 2019 Esmat E. Zein., et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.