Research Article
Volume 7 Issue 2 - 2025
Project Optimus: Principles, Challenges, and a Paradigm Shift in Dose Optimization for Cancer Therapies
University of Manchester, Faculty of Biology, Medicine and Health, School of Medical Sciences, Division of Cancer Sciences
*Corresponding Author: Hojouj M, University of Manchester, Faculty of Biology, Medicine and Health, School of Medical Sciences, Division of Cancer Sciences.
Received: February 25, 2025; Published: March 03, 2025
Abstract
Phase 1 trial designs for determining appropriate doses of cytotoxic agents have traditionally been based on the assumption that both clinical benefit and toxicity increase with higher doses. These studies aim to establish the maximum tolerated dose (MTD) for further development. However, for targeted non-cytotoxic therapies, maximum efficacy may be achieved at doses below the MTD. To address this, the FDA has introduced Project Optimus (PO) to reform the paradigm of dose optimisation and selection in cancer drug development. PO seeks to strike a balance by ensuring treatment efficacy at doses that minimise avoidable toxicities. According to PO guidance, dose escalation decisions in Phase 1 trials should incorporate preclinical data (preferably from models predicting human efficacy, toxicity, and receptor engagement), toxicity profiles (including early, delayed, low-grade toxicities, and patient-reported outcomes), pharmacokinetics (PK), pharmacodynamics (PD), and efficacy data. Rather than identifying a single dose, Phase 1 studies should determine a dose range where efficacy has been observed. The adoption of PO principles is anticipated to have a significant impact on early oncology drug development. This presentation outlines the key guidance from PO and the challenges that arise.
Background
The approach underpinning dose selection for cytotoxic chemotherapy in oncology assumes that both clinical benefit and toxicity increase with dose. Early studies aim to determine the maximum tolerated dose (MTD), which can then be used in subsequent research and ultimately in clinical practice. Traditionally, the MTD has been established using the 3+3 design, first introduced in the 1940s (Dixon and Mood, 1946). In simple terms, this method begins by administering the initial dose to three participants. If none of the three patients experiences a dose-limiting toxicity (DLT), the dose is escalated. If one of the three participants develops a DLT, three additional participants are treated at the same dose. However, if two of the three participants experience DLTs, the dose is de-escalated. Once six participants have been treated at a given dose, the following criteria apply: if 0/6 or 1/6 participants experience a DLT, the dose is escalated; if 2/6 participants experience a DLT, the dose is either de-escalated or, if six participants have already been treated at that level, the next lower dose is selected as the MTD (refer to Figure 1). Key definitions and standard parameters for this study design are outlined in Table 1 (Saxena et al., 2022).
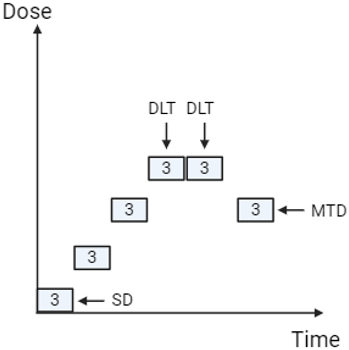
Figure 1: Traditional 3+3 design (Created with BioRender.com)
| Dose-limiting toxicities (DLTs) | Predefined toxicities (generally clinically relevant grade 3 or higher toxicities using severity criteria defined in CTCAE) that emerge during the DLT period |
| DLT period | The period during which participants are observed for DLTs, generally within 1 cycle or 3 to 4 weeks from the first exposure to the investigational agent. |
| Maximum tolerated dose (MTD) | The highest dose at which fewer than 1/3 of the participants experience DLTs. This dose is typically evaluated in later studies for chemotherapy. |
| Maximum administered dose (MAD) | The pre-agreed maximum dose is administered in a study if the MTD is not identified. |
| Recommended Phase two-dose (RP2D) | The dose is recommended for further development following a dose escalation study. This may be the MTD or a lower dose, depending on the findings. |
| Study population | Typically includes individuals who have exhausted all therapeutic options for their disease. Different tumour types are commonly included. |
| Starting dose (SD) | The initial dose administered to the first cohort in the study. This is generally a dose anticipated to provide an exposure that caused no toxicities in animals during preclinical safety studies (i.e. a fraction of the no-observed-adverse-effect level in animals). For agents where animal data may not predict human toxicity (e.g. immuno-oncology agents), the starting dose may be based on an exposure level at which the first signs of biological activity were observed in preclinical models. |
| Dose escalation increments | The magnitude of dose escalation for each new cohort may be predetermined (e.g. using a modified Fibonacci sequence with smaller increases for each new cohort). Alternatively, the magnitude of escalation may be agreed upon by the safety review committee based on emerging safety data. Bayesian statistics may also be used to support these decisions |
Table 1: Terminology for classical 3+3 dose escalation studies.
To safeguard participants’ well-being, the starting dose is set significantly below the expected efficacious dose (see Table 1). Furthermore, for first-in-class investigational agents, it is advised that the trial therapy be administered initially to a single individual who is monitored for a predefined period before the remaining participants in the cohort are enrolled. This approach, known as sentinel dosing, enhances safety.
A common criticism of the 3+3 design is that it may result in a large proportion of participants receiving sub-therapeutic doses. To address this issue, alternative approaches such as Accelerated Titration Designs (ATD) have been developed, which aim to minimise the number of participants (using cohorts of one or two) receiving doses with little or no biological effect. In an ATD, the study transitions to the classic 3+3 design once low-grade adverse events (AEs) are observed or a predefined exposure threshold is reached (see Figure 2) (Simon et al., 1997).
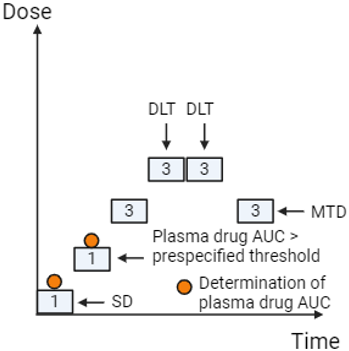
Figure 2: Accelerated Titration Design Example (Created with BioRender.com)
Dose decision-making based solely on predefined dose-limiting toxicities (DLTs) during a specified DLT period—typically 21 or 28 days—can fail to account for toxicities that arise later or for lower-grade but troublesome toxicities, such as Grade 2 diarrhoea. Moreover, identifying a maximum tolerated dose (MTD) may not always be appropriate, particularly when efficacy is evident without the occurrence of DLTs. Bayesian statistical methods offer an alternative approach to dose escalation decisions by incorporating tolerability data alongside DLTs and improving predictions of toxicity (Kurzrock et al., 2021).
In clinical trials, therapeutic and/or biological efficacy is typically evaluated using radiological imaging, liquid biopsies (e.g., blood or plasma), or tumour biopsies. The concept of biologically effective doses is well-recognised, and toxicity data combined with efficacy findings can be used to determine a recommended phase two dose (RP2D). Many dose escalation studies now adopt Bayesian statistical approaches, which are frequently used to identify and report RP2D outcomes (Hansen et al., 2017).
What Data is needed to Characterise Doses for Further Development?
For cytotoxic chemotherapy agents with a narrow Therapeutic Index (TI) (see Figure 3), it is generally appropriate to develop these agents at the maximum tolerated dose (MTD). However, over the past few decades, a deeper understanding of tumour biology and the interactions between tumours and the immune system has led to unprecedented growth in novel classes of agents for cancer treatment. These include molecularly targeted small molecules such as tyrosine kinase inhibitors, immune-oncology therapies like immune checkpoint inhibitors (ICIs), and more recently, cell-based approaches such as CAR-T therapies
For cytotoxic chemotherapy agents with a narrow Therapeutic Index (TI) (see Figure 3), it is generally appropriate to develop these agents at the maximum tolerated dose (MTD). However, over the past few decades, a deeper understanding of tumour biology and the interactions between tumours and the immune system has led to unprecedented growth in novel classes of agents for cancer treatment. These include molecularly targeted small molecules such as tyrosine kinase inhibitors, immune-oncology therapies like immune checkpoint inhibitors (ICIs), and more recently, cell-based approaches such as CAR-T therapies
(Falzone, Salomone and Libra, 2018)
For new targeted agents in oncology with wider therapeutic windows, the maximal clinical benefit may be observed at doses lower than the MTD. New molecularly targeted agents, biologics, and immunotherapies often saturate their targets at doses below the MTD, indicating that lower doses may provide comparable efficacy while reducing the toxicity burden (Murphy, Halford and Stefan Nicholas Symeonides, 2023).
For new targeted agents in oncology with wider therapeutic windows, the maximal clinical benefit may be observed at doses lower than the MTD. New molecularly targeted agents, biologics, and immunotherapies often saturate their targets at doses below the MTD, indicating that lower doses may provide comparable efficacy while reducing the toxicity burden (Murphy, Halford and Stefan Nicholas Symeonides, 2023).
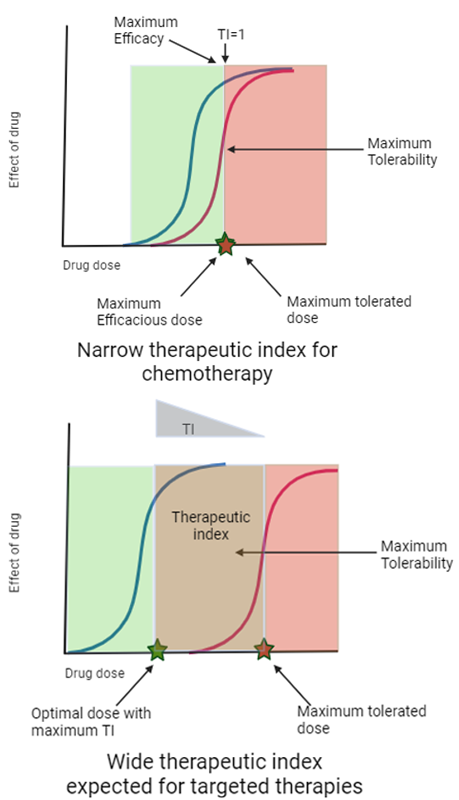
Figure 3: Illustration of potential difference in therapeutic indices (TIs) for chemotherapy & MTA (molecular targeted agents) in oncology (Created with BioRender.com)
A significant clinical challenge associated with immunotherapy is the emergence of immune-related adverse events (irAEs), which differ markedly from the toxicities commonly seen with traditional chemotherapy. With the increasing use of immune checkpoint inhibitors (ICIs) in oncology, clinicians are encountering irAEs that can affect multiple organs, including colitis, pneumonitis, endocrinopathies, liver toxicity, and nephritis. These toxicities can arise months after treatment initiation, may be life-threatening, and often require a multidisciplinary approach for effective management (Martins et al., 2019).
Unlike many chemotherapies, which are administered in limited courses, these newer agents are typically given until disease progression. As a result, the long-term benefits of treatment are accompanied by late-onset toxicities and persistent low-grade toxicities, which are increasingly significant issues, often leading to dose interruptions or therapy discontinuation (Martins et al., 2019).
The doses and schedules of several oncology therapies have required modification following regulatory approval to address safety or tolerability concerns. Notable examples include ceritinib, dasatinib, niraparib, and gemtuzumab ozogamicin (Shah et al., 2021).
The FDA has raised concerns that current methods for determining appropriate doses for new agents are often inadequate. It stated, “Too often, the current paradigm for dose selection—based on cytotoxic chemotherapeutics—results in doses and schedules for molecularly targeted therapies that are inadequately characterised before initiating registration trials.” In response, the FDA established Project Optimus to promote a new dose-finding and optimisation paradigm in oncology, focusing on selecting doses that balance efficacy with safety and tolerability (U.S. Food and Drug Administration, 2023).
To support this initiative, the FDA has collaborated with regulatory agencies, academia, the pharmaceutical industry, and patients to develop principles and guidelines for optimising doses and schedules in oncology drug development. One such multidisciplinary group, the Methodology for the Development of Innovative Cancer Therapies Taskforce (MDICT), has published recommendations on this topic (Araujo et al., 2023). This article highlights key recommendations from these guidelines and explores their implications.
Key aspects from Project Optimus recommendations (Araujo et al., 2023, U.S. Food and Drug Administration, 2024)
Preclinical data should play a crucial role in informing trial design by predicting efficacious dose ranges, evaluating the impact of dose and schedule on target engagement, efficacy, and toxicity, understanding how tumour biology influences efficacy, and identifying pharmacodynamic (PD) markers to assess treatment effects. Although these considerations are already integral to drug development, the FDA seeks to formalise their inclusion. During the study design phase, sponsors are encouraged to align with regulators on how PD modelling, derived from preclinical findings, will guide dose selection.
Preclinical data should play a crucial role in informing trial design by predicting efficacious dose ranges, evaluating the impact of dose and schedule on target engagement, efficacy, and toxicity, understanding how tumour biology influences efficacy, and identifying pharmacodynamic (PD) markers to assess treatment effects. Although these considerations are already integral to drug development, the FDA seeks to formalise their inclusion. During the study design phase, sponsors are encouraged to align with regulators on how PD modelling, derived from preclinical findings, will guide dose selection.
Early dose escalation studies should aim to identify a recommended dose range (RDR) for subsequent development. Project Optimus highlights the need to explore how varying doses influence efficacy and toxicity, rather than focusing exclusively on determining a maximum tolerated dose (MTD) or a single recommended Phase 2 dose (RP2D). The recommended dose (RD) may vary depending on the disease, tumour site (as certain sanctuary sites may require higher doses), or specific molecular alterations (e.g., the dose of imatinib differs by indication). Determining an MTD, where possible, can still provide valuable information for managing overdose scenarios or drug-drug interactions (DDIs) that increase exposure.
Decisions regarding dose escalation should take into account all available safety and tolerability data, alongside pharmacokinetics (PK), efficacy, and biological findings.
- In addition to reviewing dose-limiting toxicities (DLTs), it is important to consider adverse events (AEs) reported beyond the DLT period (late toxicities), lower-grade toxicities, and any necessary dose interruptions or reductions at any time.
- It is recommended that patient-reported outcomes (PRO) data be collected where possible. There are validated quality of life (QOL) questionnaires, some of which are general, while others are designed to collect detailed information on specific aspects (e.g., pain, fatigue, diarrhoea).
- All available efficacy and pharmacodynamic data should be reviewed. Classical tumour shrinkage (RECIST 1.1) using radiological imaging remains the gold standard. Additional imaging approaches, such as radiomics and PET CT, can provide valuable insights.
- Pharmacodynamic (PD) biomarkers may indicate biological effects specifically developed for that agent in preclinical models (e.g., evidence of pathway disruption in tumour biopsies or surrogate tissue) or may reflect a general impact on the tumour (e.g., changes in circulating tumour DNA [ctDNA]).
- Real-time PK data for all participants should be available for each dose escalation decision. Relationships between dose/exposure and efficacy, as well as dose/exposure and toxicity, should be reviewed.
It is advised that at least two dose levels be compared by randomising participants into two distinct arms to effectively assess efficacy, tolerability, and safety. The higher dose may include the maximum tolerated dose (MTD), while evidence of clinical activity should be provided for the chosen lower dose(s). Additionally, the pharmacokinetic (PK) overlap between the dose levels should be minimised. The trial does not need to be powered to show superiority or non-inferiority but should be appropriately sized to allow for a comprehensive evaluation of safety and anti-tumour activity at each dose level. This comparison can be carried out within the dose escalation study (by adding backfill cohorts) or, ideally, in a separate Phase II study. However, it is acknowledged that such randomised studies may not be feasible (for example, in the case of very rare diseases), may not be necessary (for agents with a known narrow therapeutic index, like chemotherapies), or where clear efficacy is observed in a homogeneous population with oncogene-addicted tumours. Two options for collecting dose-ranging data are illustrated in Figures 4 and 5.
The parameters observed in Project Optimus are represented in Table 2.
| Parameter | Explanation |
| Treatment Limiting toxicity (TLT) | Includes chronic low-grade toxicity, late-emerging toxicities, and non-dose-dependent toxicity that may limit the duration of therapy. |
| Recommended dose range (RDR) | The range of doses identified in the dose escalation study to be tested in a randomized setting |
| Recommended dose (RD) | The dose recommended for later-phase trials is identified through dose-ranging or dose-confirmation studies. |
| Minimal reproducible active dosage (MRAD) | Lowest dose where there is evidence of clinical activity |
Table 2: Definition and terminology utilised in Project Optimus.
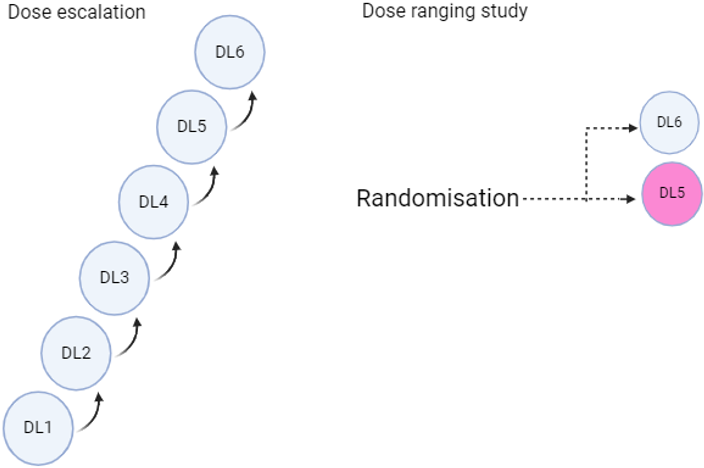
Figure 4: Dose finding using separate dose escalation & dose ranging studies (Created with BioRender.com)
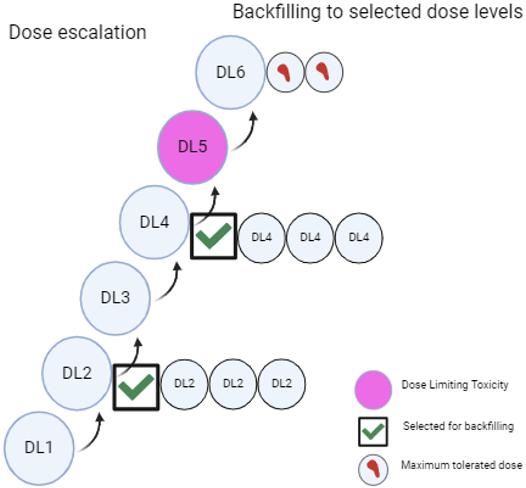
Figure 5: Dose finding through backfilling to cohorts in the dose escalation study (Created with BioRender.com)
Implications for the Project Optimus Recommendations
When examining certain individual elements within the guidelines, demonstrating differences in efficacy between doses with a small patient sample is likely to be challenging. Response rates, such as tumour shrinkage measured radiologically using RECIST 1.1, serve as an efficacy endpoint often utilised in single-arm studies to support approval for some oncology drugs. However, cancer drugs are typically approved based on comparisons with standard care therapies in Phase III randomised studies. Overall survival (OS), regarded as the gold standard, and progression-free survival (PFS) are the primary regulatory endpoints. To show a statistically significant and clinically meaningful improvement in OS and/or PFS, large-scale studies are required. There may be a discrepancy between response rates and PFS/OS outcomes. For instance, the Phase III Confirm study assessed two dose levels of fulvestrant (250mg and 500mg intramuscularly, monthly) in metastatic HR+ breast cancer. In this study, which included over 700 patients, the objective response rate was 9.1% at the 500mg dose and 10.2% at the 250mg dose, but the PFS was more favourable for the 500mg dose (Hazard Ratio (HR) 0.80 (0.68-0.94)) (Angelo Di Leo et al., 2010). In this case, the difference in response rates did not reflect the more clinically significant difference in PFS.
When examining certain individual elements within the guidelines, demonstrating differences in efficacy between doses with a small patient sample is likely to be challenging. Response rates, such as tumour shrinkage measured radiologically using RECIST 1.1, serve as an efficacy endpoint often utilised in single-arm studies to support approval for some oncology drugs. However, cancer drugs are typically approved based on comparisons with standard care therapies in Phase III randomised studies. Overall survival (OS), regarded as the gold standard, and progression-free survival (PFS) are the primary regulatory endpoints. To show a statistically significant and clinically meaningful improvement in OS and/or PFS, large-scale studies are required. There may be a discrepancy between response rates and PFS/OS outcomes. For instance, the Phase III Confirm study assessed two dose levels of fulvestrant (250mg and 500mg intramuscularly, monthly) in metastatic HR+ breast cancer. In this study, which included over 700 patients, the objective response rate was 9.1% at the 500mg dose and 10.2% at the 250mg dose, but the PFS was more favourable for the 500mg dose (Hazard Ratio (HR) 0.80 (0.68-0.94)) (Angelo Di Leo et al., 2010). In this case, the difference in response rates did not reflect the more clinically significant difference in PFS.
Will higher, potentially more effective doses be prematurely eliminated in early development due to a lack of significant differences in surrogate outcomes such as overall response rate, duration of responses, or changes in PD biomarkers?
To address this concern, the clinical community is working to validate alternative endpoints beyond those included in RECIST 1.1, using novel imaging techniques such as radiomics and PET scans to assess responses (E de Vries, 2024). Additionally, there is an ongoing initiative to standardise approaches for measuring ctDNA and define responses based on ctDNA changes (Garralda, 2024). Data from current and future early studies will help validate these novel efficacy endpoints. However, it is important to recognise that it will take time to determine whether changes in these dynamic markers can predict better long-term clinical outcomes and eventually become new standard endpoints.
PD biomarkers, used to demonstrate biological effects, must be validated and discussed with the FDA before study initiation.
Many approaches for evaluating patient-reported outcome (PRO) data are still in the early stages of development and will need to be carefully considered when integrating them into dose escalation studies. Typically, dose escalation studies involve participants who have already exhausted standard care therapies, often resulting in very heterogeneous populations. Factors such as the sites of metastases, disease burden, and the number and type of prior treatments can vary widely. These factors may significantly affect a patient’s symptoms, comorbidities, and quality of life, independent of the investigational agent’s effect, making the interpretation of data challenging, especially when cohort sizes are small. Furthermore, appropriate health-related quality of life (HRQOL) instruments need to be validated for early clinical development, considering the time patients, often with limited life expectancy, must invest in participating in a clinical trial.
Project Optimus introduces additional complexities in the design of early clinical trials for oncology drug development, which affects both the speed and initial costs of drug development. Smaller companies are likely to feel the impact most, given their limited resources, particularly funding, with pressure from investors to deliver clinical results as quickly as possible.
On the positive side, many of the principles in the Project Optimus guidelines have already been part of early clinical drug design for years. These include the use of preclinical data modelling, Bayesian model-based designs, simulations, more sophisticated dose escalation decisions that incorporate long-term safety and tolerability data, and PK-PD relationships. Project Optimus formalises and explicitly outlines the dose escalation decision-making process, typically handled by the Safety or Cohort Review Committee (SRC/CRC).
Moreover, Project Optimus is not merely a set of guidelines for early study designs; it represents a fundamental shift in the philosophy of identifying appropriate doses throughout all stages of oncology drug development. It fosters a more collaborative approach between drug developers and regulatory bodies. Dose optimisation plans require early engagement with regulatory authorities as part of clinical study design discussions and may be revisited during milestone meetings. The FDA has indicated that discussions on dose-finding strategies should not be limited to milestone meetings; separate meetings may be necessary as new clinical data becomes available. This collaborative approach aims to determine the best optimal dose and schedule for patients, based on emerging risk/benefit data from explored doses and schedules, before the drug progresses into Phase III trials and eventually reaches cancer patients in the clinic.
1Division of Cancer Sciences- The University of Manchester, UK
2Simbec-Orion Ltd, London, UK.
3DeLondra Oncology Ltd, UK.
4Division of Clinical Medicine, the University of Sheffield, UK
5Kingston Oncology Ltd, UK
1Division of Cancer Sciences- The University of Manchester, UK
2Simbec-Orion Ltd, London, UK.
3DeLondra Oncology Ltd, UK.
4Division of Clinical Medicine, the University of Sheffield, UK
5Kingston Oncology Ltd, UK
References
- American Society of Clinical Oncology Educational Book. (2021). Moving Beyond 3+3: The Future of Clinical Trial Design | American Society of Clinical Oncology Educational Book. [online] Available at: https://ascopubs.org/doi/pdf/10.1200/EDBK_319783 [Accessed 27 Sep. 2024].
- Angelo Di Leo, Jerusalem, G., Lubos Petruzelka, Torres, R., Bondarenko, I.N., Rustem Khasanov, Verhoeven, D., Pedrini, J.L., Smirnova, I., Lichinitser, M.R., Pendergrass, K., Garnett, S., Justin P.O. Lindemann, Sapunar, F. and Martin, M. (2010). Results of the CONFIRM Phase III Trial Comparing Fulvestrant 250 mg With Fulvestrant 500 mg in Postmenopausal Women With Estrogen Receptor–Positive Advanced Breast Cancer. Journal of Clinical Oncology, [online] 28(30): 4594–4600.
- Araujo, D., Greystoke, A., Bates, S., Bayle, A., Calvo, E., Castelo-Branco, L., de Bono, J., Drilon, A., Garralda, E., Ivy, P., Kholmanskikh, O., Melero, I., Pentheroudakis, G., Petrie, J., Plummer, R., Ponce, S., Postel-Vinay, S., Siu, L., Spreafico, A. and Stathis, A. (2023). Oncology phase I trial design and conduct: time for a change - MDICT Guidelines 2022. Annals of Oncology, [online] 34(1): 48–60.
- Dixon, W.J. and Mood, A.M. (1946). The Statistical Sign Test. Journal of the American Statistical Association, 41(236): 557–566.
- Falzone, L., Salomone, S. and Libra, M. (2018). Evolution of Cancer Pharmacological Treatments at the Turn of the Third Millennium. Frontiers in Pharmacology, [online] 9.
- Hansen, A.R., Cook, N., Amir, E., Siu, L.L. and Abdul, R. (2017). Determinants of the recommended phase 2 dose of molecular targeted agents. Cancer, [online] 123(8): 1409–1415.
- Garralda E., Incorporating ctDNA into RECIST v1.1: Steps ahead. Presentation at ESMO Congress Sept 2024
- Martins, F., Latifyan Sofiya, Sykiotis, G.P., Faiza Lamine, Maillard, M., Fraga, M., Keyvan Shabafrouz, Camillo Ribi, Cairoli, A., Guex-Crosier, Y., Thierry Kuntzer, Olivier Michielin, Peters, S., Georges Coukos, Francois Spertini, Thompson, J.A. and Obeid, M. (2019). Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nature Reviews Clinical Oncology, [online] 16(9): 563–580.
- Murphy, R., Halford, S. and Stefan Nicholas Symeonides (2023). Project Optimus, an FDA initiative: Considerations for cancer drug development internationally, from an academic perspective. Frontiers in Oncology.
- Saxena, A., Rubens, M., Ramamoorthy, V., Zhang, Z., Md Ashfaq Ahmed, McGranaghan, P., Das, S. and Emir Veledar (2022). A Brief Overview of Adaptive Designs for Phase I Cancer Trials. Cancers, [online] 14(6): 1566–1566.
- Shah, M., Rahman, A., Theoret, M.R. and Pazdur, R. (2021). The Drug-Dosing Conundrum in Oncology — When Less Is More. New England Journal of Medicine. 385(16): 1445–1447.
- Simon, R., Rubinstein, L., Arbuck, S.G., Christian, M.C., Freidlin, B. and Collins, J. (1997). Accelerated Titration Designs for Phase I Clinical Trials in Oncology. JNCI Journal of the National Cancer Institute, 89(15): 1138–1147.
- U.S. Food and Drug Administration, 2023 https://www.fda.gov/about-fda/oncology-center-excellence/project-optimus
- U.S. Food and Drug Administration, 2024 https://www.fda.gov/regulatory-information/search-fda-guidance-documents/optimizing-dosage-human-prescription-drugs-and-biological-products-treatment-oncologic-diseases
- Vries E., Innovative imaging techniques to evaluate response. Presentation at ESMO Congress Sept (2024).
Citation: Hojouj M, Landers D, Cruz R, Clack G and Stuart M. (2025). Project Optimus: Principles, Challenges, and a Paradigm Shift in Dose Optimization for Cancer Therapies. Journal of Pharmacy and Drug Development 7(2). DOI: 10.5281/zenodo.15016057
Copyright: © 2025 Hojouj M. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.