Research Article
Volume 2 Issue 1 - 2020
Spectrophotometric Method Development and Validation for the Determination of Losartan Potassium in Pharmaceutical Formulations using Hydroxypropyl-β-cyclodextrin
Chemistry Department, Faculty of Science, University of Khartoum, P.O. Box 321, Khartoum, Sudan
*Corresponding Author: Prof. Abdalla Ahmed Elbashir, Chemistry Department, Faculty of Science,University of Khartoum, Khartoum, Sudan.
Received: June 06, 2020; Published: July 08, 2020
Abstract
A new, simple, and sensitive spectrophotometric method has been developed for the determination of Losartan Potassium based on the host-guest inclusion reaction with hydroxypropyl-β-cyclodextrin reagent. The maximum absorbance intensity of Losartan potassium was found to be at 205 nm whereas for the complex at 208 nm with a slight shift in the wavelength. The inclusion product was practically examined at optimum pH 1.0. Furthermore, the stoichiometry was found to be 1:1 and it was characterized using FT-IR spectrometer by comparing the obtainable spectra. We can obviously notice the disappearance of the secondary amine group peak and the CH aliphatic, methyl, and alkyl halide groups peaks from the Losartan potassium spectrum, which they have been entrapped into the Hydroxylpropyl-β-cyclodextrin reagent cavity due to the hydrogen bond formation and resulting in a significant change in the hydroxyl group peak intensity from 3392 cmc-1 to 3346 cm-1 and shape. In addition to that, validation of the proposed method was clearly interpreted by the obtainable linearity over the concentration range of 1.0-10 mgLc-1 with good correlation coefficient (R2) 0.9997, the limits of detection (LOD) and quantification (LOQ) were obtained 0.1327 and 0.4022 µgmlc-1 respectively. All parameters have been calculated as per International Conference on Harmonization (ICH) guidelines. The developed method is successfully applicable for the determination of Losartan potassium in pharmaceutical dosage formulations.
Keywords: Losartan potassium; UV spectrophotometer; Hydroxypropyl-β-cyclodextrin; Inclusion complex; FT-IR.
Introduction
Losartan potassium (LoP) Figure 1 is an angiotensin ?? receptor antagonists drug used primarily in the treatment of high blood pressure (hypertension), to reduce the risk of stroke and help kidneys to work properly especially with diabetic patients. Physically, LoP is a white crystalline powder, highly soluble in water and alcohol. Moreover, LoP chemically is 2-butyl-4-chloro-1-[p-(o-1H-tetrazol-5-ylphenyl)benzyl]imidazole-5-methanol mono potassium salt[1-5]. Various analytical methods for the determination of LoP in pharmaceutical formulations have been reported such as HPLC, high performance thin layer chromatography, super-critical fluid chromatography [6-8], UV and derivative UV spectrophotometry, and capillary electrophoresis [9-12]. Additionally, the only supramolecular interaction with LoP was carried out using ESI mass-spectrometry and was characterized by NMR technique [13]. However, some of these methods are time consuming, costly, and tedious. Hence a reasonable demand for a rapid, simple, sensitive, and economical procedure is noticeable, especially for the routine analysis of pharmaceutical drugs that containing LoP. Consequently, in this work a development of a UV spectrophotometric method for the inclusion complex between LoP and cyclodextrin had been successfully accomplished. When it comes to talk about cyclodextrins (CDs), let us define the CDs concept which is known as a cyclic oligosaccharides consisting of glycopyranose units in truncated cone shape having lipophilic central cavity and hydrophilic outer surface. There are three natural types of CDs; α-,β-, and γ-cyclodextrins which contain six, seven, and eight glycopyranose units respectively [14-16]. CDs can readily form inclusion complexes with guest molecules by either fully or partially entrapping into their cavity, this inclusion formation depends mainly on significant factors such like polarity, hydrophobicity, shape, and size of the guest. And as a result of that, the guest`s physiochemical properties can greatly be improved just like solubility, reactivity, and stability. Hosting of drugs molecules occurred by intermolecular hydrogen bonds due to the presence of the hydroxyl groups on the outer surface of the CDs forming water soluble host-guest complexes [17-22]. Thence these positive effects have been widely applied to the remarkable improvement of the pharmaceuticals stability and solubility in the pharmaceutical industry field. In addition to that, CDs have a wide range of practical application in pharmaceutical [23, 24], cosmetic, food chemical [25, 26], and other industries. It uses for a various purposes which include solubility enhancement of insoluble substances, stabilization of labile guests against oxidation, light and heat, control of volatility and controlled release of drugs, flavors and various branches of analytical chemistry[27-29]. From an analytical perspective the formation of inclusion complexes allows to enhance fluorescence intensity [30-33] and induce chiral separation in capillary electrophoresis (CE)[34-37] have made comprehensive review on this topic for determination of several pharmaceutical drugs, pesticides, and metal ion [38].

Figure 1: Losartan Potassium chemical structure.
Recently, many applications of CDs have been effectively exploited particularly hydroxypropyl-β-cyclodextrin Figure 2 and that was justified because of its higher water solubility, greater biocompatibility possession, and its lowest toxicity [39, 40]. The aim of this work was to develop a rapid, sensitive, and economical UV spectrophotometric method for the determination of LoP in pharmaceutical dosage formulations using hydroxypropyl-β-cyclodextrin reagent.

Figure 2: Hydroxylpropyl-β-cyclodextrin (HPβCD).
Experimental Materials and Methods
Chemicals and materials
Losartan potassium (LoP) standard was received from Blue Nile Pharmaceuticals Factory, Khartoum, Sudan. Hydroxylpropyl-β-cyclodextrin (HPβCD) was obtained from Sigma-Aldrich (ST. Louis, USA) and used without prior treatment. The two pharmaceutical formulations Nilozaar and Amilosan (50mg/tablet) were purchased from the local market, which were manufactured by Blue Nile Pharmaceuticals Factory, Sudan and Amipharma Laboratories Ltd. Sudan respectively. Doubled distilled water was used in all experiments and all reagents used were analytical grade.
Losartan potassium (LoP) standard was received from Blue Nile Pharmaceuticals Factory, Khartoum, Sudan. Hydroxylpropyl-β-cyclodextrin (HPβCD) was obtained from Sigma-Aldrich (ST. Louis, USA) and used without prior treatment. The two pharmaceutical formulations Nilozaar and Amilosan (50mg/tablet) were purchased from the local market, which were manufactured by Blue Nile Pharmaceuticals Factory, Sudan and Amipharma Laboratories Ltd. Sudan respectively. Doubled distilled water was used in all experiments and all reagents used were analytical grade.
Instruments
A PharmaSpec UV-1700 ultra violet spectrophotometer supplied with 1-cm quartz cell, Shimadzu, Japan, has been used for all the quantitative measurement. pH meter model pH-3E for adjusting the pH. FT-IR data was investigated by Shimadzu FT-IR-8400S Fourier transform infrared spectrophotometer.
A PharmaSpec UV-1700 ultra violet spectrophotometer supplied with 1-cm quartz cell, Shimadzu, Japan, has been used for all the quantitative measurement. pH meter model pH-3E for adjusting the pH. FT-IR data was investigated by Shimadzu FT-IR-8400S Fourier transform infrared spectrophotometer.
Preparations of standard and sample solutions
Preparation of Hydroxylpropyl-β-cyclodextrin (HPβCD) reagent
A 0.004 molL-1 stock solution was prepared by dissolving 0.5501 g in 100 ml calibrated flask of doubled distilled water.
A 0.004 molL-1 stock solution was prepared by dissolving 0.5501 g in 100 ml calibrated flask of doubled distilled water.
Preparation of Losartan potassium (LoP)
A 100 mgL-1 Stock solution was prepared by dissolving 0.01g in 100 ml standard flask of doubled distilled water.
A 100 mgL-1 Stock solution was prepared by dissolving 0.01g in 100 ml standard flask of doubled distilled water.
Preparation of phosphate buffer solutions
A 0.1 molL-1 phosphate buffer solution was prepared by dissolving appropriate amounts of sodium dihydrogen phosphate (NaH2PO4) and disodium hydrogen phosphate (Na2HPO4) in doubled distilled water, the pH was adjusted from 1.0 to 7.0 using 0.1 molL-1 hydrochloric acid (HCl) solution.
A 0.1 molL-1 phosphate buffer solution was prepared by dissolving appropriate amounts of sodium dihydrogen phosphate (NaH2PO4) and disodium hydrogen phosphate (Na2HPO4) in doubled distilled water, the pH was adjusted from 1.0 to 7.0 using 0.1 molL-1 hydrochloric acid (HCl) solution.
General recommended procedure
A 0.6 ml of 100 mgL-1LoP was transferred into a 10.00 ml volumetric flask, 3.0 ml of 0.1 molL-1 phosphate buffer solution of pH 1.0, and 1.0 ml of 4.0×10-2 molL-1 HPβCD were added sequentially, then diluted to the mark with doubled distilled water and mixed well. Having equilibrated the solution at 35?C for 5 min, the absorbance was measured at 208 nm against blank reagent prepared with the same reagents concentrations except the drug. The same method was applied to obtain the linear calibration graph by using aliquot volumes of LoP working solutions equivalent to 1.0-10 mgL-1, then following the general procedure and plotting the absorbance versus the concentration of drug in mgL-1.
A 0.6 ml of 100 mgL-1LoP was transferred into a 10.00 ml volumetric flask, 3.0 ml of 0.1 molL-1 phosphate buffer solution of pH 1.0, and 1.0 ml of 4.0×10-2 molL-1 HPβCD were added sequentially, then diluted to the mark with doubled distilled water and mixed well. Having equilibrated the solution at 35?C for 5 min, the absorbance was measured at 208 nm against blank reagent prepared with the same reagents concentrations except the drug. The same method was applied to obtain the linear calibration graph by using aliquot volumes of LoP working solutions equivalent to 1.0-10 mgL-1, then following the general procedure and plotting the absorbance versus the concentration of drug in mgL-1.
Assay procedure for Losartan potassium (LoP) tablets
Twenty tablets were carefully weighed and finely powdered. An accurate weight equivalent to 50 mg of LoP was transferred into 100 ml conical flask and instantly dissolved and extracted in 60 ml doubled distilled water. The residue was filtered through Whatmann filter paper no. 41 then transferred into 100 ml standard flask and diluted to the mark to obtain 100 mgL-1 stock solution. Correspondingly, the prepared solutions were pursued the general recommended procedure.
Twenty tablets were carefully weighed and finely powdered. An accurate weight equivalent to 50 mg of LoP was transferred into 100 ml conical flask and instantly dissolved and extracted in 60 ml doubled distilled water. The residue was filtered through Whatmann filter paper no. 41 then transferred into 100 ml standard flask and diluted to the mark to obtain 100 mgL-1 stock solution. Correspondingly, the prepared solutions were pursued the general recommended procedure.
Stoichiometric ratio of the inclusion reaction (Job`s method)
The stoichiometry determination of the inclusion complex was successfully performed using Job`s method of continuous variation [41]. Equimolar 1.0×10-4 molL-1aqueous solutions of LoP and HPβCD were prepared, then a series of 10.00 ml portions were made up constituting various complementary proportions (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1 ml). These solutions were properly treated as described in the general recommended procedure.
The stoichiometry determination of the inclusion complex was successfully performed using Job`s method of continuous variation [41]. Equimolar 1.0×10-4 molL-1aqueous solutions of LoP and HPβCD were prepared, then a series of 10.00 ml portions were made up constituting various complementary proportions (1:9, 2:8, 3:7, 4:6, 5:5, 6:4, 7:3, 8:2, and 9:1 ml). These solutions were properly treated as described in the general recommended procedure.
Characterization of LoP/HPβCD product
The formed complex was investigated using Shimadzu FT-IR-8400S Fourier transform infrared spectrometer (FT-IR). The diffuse reflectance technique was carried out in the spectral region of 400-4000 cm-1. The procedure consisting of placing the powdered dried sample which dispersed in KBr with ratio of 1:100 respectively into a sampling cup, smoothing the powder into a disc, and compressing the powder using hand compressor. Having placed the sample into the light path, the spectrum was obtained. The FT-IR spectra of pure LoP, HPβCD, and LoP/HPβCD complex were also obtained following the same described procedure for comparison.
The formed complex was investigated using Shimadzu FT-IR-8400S Fourier transform infrared spectrometer (FT-IR). The diffuse reflectance technique was carried out in the spectral region of 400-4000 cm-1. The procedure consisting of placing the powdered dried sample which dispersed in KBr with ratio of 1:100 respectively into a sampling cup, smoothing the powder into a disc, and compressing the powder using hand compressor. Having placed the sample into the light path, the spectrum was obtained. The FT-IR spectra of pure LoP, HPβCD, and LoP/HPβCD complex were also obtained following the same described procedure for comparison.
Results and Discussion
Absorption spectra
The absorption spectra of LoP in the presence and absence of HPβCD reagent was carefully studied in the range of 200-400 nm. The obtainable results as shown in Figure 3 had revealed that the wavelength of the maximum absorbance of 6.0 mgL-1LoP was found to have Lambdamaximum at 205 nm. When 4.0×10-4 molL-1 HPβCD was added to 6.0 mgL-1LoP in a buffer solution of pH 1.0, we clearly observed theconsiderable increase of the absorbance and the slight shift in the wavelengthinto 208 nm. This experimentally confirmed the capability of LoP to form inclusion complex with HPβCD reagent.
The absorption spectra of LoP in the presence and absence of HPβCD reagent was carefully studied in the range of 200-400 nm. The obtainable results as shown in Figure 3 had revealed that the wavelength of the maximum absorbance of 6.0 mgL-1LoP was found to have Lambdamaximum at 205 nm. When 4.0×10-4 molL-1 HPβCD was added to 6.0 mgL-1LoP in a buffer solution of pH 1.0, we clearly observed theconsiderable increase of the absorbance and the slight shift in the wavelengthinto 208 nm. This experimentally confirmed the capability of LoP to form inclusion complex with HPβCD reagent.

Figure 3: Absorption spectra of HPβCD (4.0×10-4 molL-1) against water (?) LoP against water (?), and the reaction product of LoP (6.0 mgL-1) with HPβCD (4.0×10-4 molL-1) against the reagent blank (?).
FT-IR characterization of inclusion complex
The IR spectrum of LoP/HPβCD product was conducted over the wave number from 400-4000 cm-1 using FT-IR spectrometer as described in Figure 4. The IR spectrum of LoP was characterized by the presence of main peaks for secondary amine group, hydroxyl group, CH aliphatic and CH aromatic groups, substituted amine –N–K group, methyl CH3 group, and alkyl halide C–Cl group. Alternatively, the presence of a strong stretching peak of hydroxyl group related to pure HPβCD reagent. In the spectrum, not only had the peaks shifted, but also some of them disappeared specifically when comparing the spectra of LoP and the obtainable complex. LoP showed different peaks at 3614, 3188, 2945, and 2864 cm-1 for secondary amine stretching group, hydroxyl aromatic stretching group, CH aromatic stretching group, and CH aliphatic bending group. Also,at 1662 and 1573 cm-1 corresponding to the C=C alkene bending group and –N–K stretching group, while at 1458 and 1421 cm-1 for the methyl stretching group and C=C alkene bending group. The nitrile CN groups peaks at 1255, 1195, and 999 cm-1. The alkyl halide C–Cl group at 757 cm-1[42-44]. When analyzing the FT-IR spectra of LoP, HPβCD, and LoP/HPβCD we can recognize the change and shifts in the CH aromatic group at 2939 cm-1, nitrile group at 2339 cm-1, alkene group at 1668 cm-1, amine group at 1533cm-1, and nitrile groups at 1274, 1139, and 991 cm-1 which indicated these groups were not changed after the inclusion complex formation. Nevertheless, the disappearance of the secondary amine group due to the formation of hydrogen bond and that because of the presence of the hydroxyl group. Likewise, the disappearance of the other functional groups as CH aliphatic, methyl, and alkyl halide groups due to the entrapment into the HPβCD cavity. Only once the LoP had been entrapped into the HPβCD cavity was a dramatic transformation in the HPβCD hydroxyl group peak shape and intensity inspected from 3392 cm-1 into 3346 cm-1 which clearly interpreted the formation of LoP/HPβCD inclusion complex Figure 5.
The IR spectrum of LoP/HPβCD product was conducted over the wave number from 400-4000 cm-1 using FT-IR spectrometer as described in Figure 4. The IR spectrum of LoP was characterized by the presence of main peaks for secondary amine group, hydroxyl group, CH aliphatic and CH aromatic groups, substituted amine –N–K group, methyl CH3 group, and alkyl halide C–Cl group. Alternatively, the presence of a strong stretching peak of hydroxyl group related to pure HPβCD reagent. In the spectrum, not only had the peaks shifted, but also some of them disappeared specifically when comparing the spectra of LoP and the obtainable complex. LoP showed different peaks at 3614, 3188, 2945, and 2864 cm-1 for secondary amine stretching group, hydroxyl aromatic stretching group, CH aromatic stretching group, and CH aliphatic bending group. Also,at 1662 and 1573 cm-1 corresponding to the C=C alkene bending group and –N–K stretching group, while at 1458 and 1421 cm-1 for the methyl stretching group and C=C alkene bending group. The nitrile CN groups peaks at 1255, 1195, and 999 cm-1. The alkyl halide C–Cl group at 757 cm-1[42-44]. When analyzing the FT-IR spectra of LoP, HPβCD, and LoP/HPβCD we can recognize the change and shifts in the CH aromatic group at 2939 cm-1, nitrile group at 2339 cm-1, alkene group at 1668 cm-1, amine group at 1533cm-1, and nitrile groups at 1274, 1139, and 991 cm-1 which indicated these groups were not changed after the inclusion complex formation. Nevertheless, the disappearance of the secondary amine group due to the formation of hydrogen bond and that because of the presence of the hydroxyl group. Likewise, the disappearance of the other functional groups as CH aliphatic, methyl, and alkyl halide groups due to the entrapment into the HPβCD cavity. Only once the LoP had been entrapped into the HPβCD cavity was a dramatic transformation in the HPβCD hydroxyl group peak shape and intensity inspected from 3392 cm-1 into 3346 cm-1 which clearly interpreted the formation of LoP/HPβCD inclusion complex Figure 5.

Figure 4: FT-IR spectrum (?) HPβCD, (?) LoP, and (?) LoP/HPβCD complex.

Figure 5: Formed LoP/HPβCD inclusion complex.
Optimization of the reaction conditions
The inclusion reaction of LoP and HPβCD reagent greatly affected by various experimental parameters just like the pH, volume of buffer, concentration of reagent, temperature, and reaction time that were intensively studied and optimized.
The inclusion reaction of LoP and HPβCD reagent greatly affected by various experimental parameters just like the pH, volume of buffer, concentration of reagent, temperature, and reaction time that were intensively studied and optimized.
Effect of pH
The influence of pH on the absorption intensity of LoP/HPβCD complex was investigated over the range 1.0 –7.0. The acidity of the solution was carefully adjusted by phosphate buffer solution. As shown in Figure 6, the maximum absorbance was noticed at pH 1.0 and started to gradually decrease until pH 7.0. Moreover, LoP is considered to be acidic due to the presence of the protonated secondary amine groups, so pH 1.0 had been reported as the optimum pH and properly chosen for the inclusion reaction with HPβCD.
The influence of pH on the absorption intensity of LoP/HPβCD complex was investigated over the range 1.0 –7.0. The acidity of the solution was carefully adjusted by phosphate buffer solution. As shown in Figure 6, the maximum absorbance was noticed at pH 1.0 and started to gradually decrease until pH 7.0. Moreover, LoP is considered to be acidic due to the presence of the protonated secondary amine groups, so pH 1.0 had been reported as the optimum pH and properly chosen for the inclusion reaction with HPβCD.

Figure 6: Effect of buffer pH on the reaction of LoP (6.0 mgL-1) with HPβCD.
Effect of volume of buffer
The influence of volume of phosphate buffer on the absorbance of the reaction product was studied using different volumes. It was found that the absorbance of LoP/HPβCD complex began to increase from 1.5 to 3.0 ml then dramatically decreased as shown in Figure 7. Therefore, 3.0 ml of phosphate buffer of pH 1.0 had been selected as optimal volume.
The influence of volume of phosphate buffer on the absorbance of the reaction product was studied using different volumes. It was found that the absorbance of LoP/HPβCD complex began to increase from 1.5 to 3.0 ml then dramatically decreased as shown in Figure 7. Therefore, 3.0 ml of phosphate buffer of pH 1.0 had been selected as optimal volume.
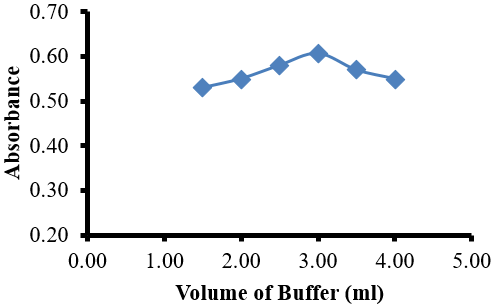
Figure 7: Effect of volume of buffer (pH 1.0) on the reaction of LoP (6.0 mgL-1) with HPβCD.
Effect of concentration of HPβCD reagent
The effect of HPβCD concentration was examined in intervals of (1.0 – 5.0) ×10-4 molL-1. It was observed that increasing in the concentration of HPβCD reagent caused a noticeable increase in the product absorbance when reaching 4.0×10-4 molL-1 as shown in Figure 8. Accordingly, 4×10-4 molL-1 of HPβCD reagent had been chosen as optimal concentration.
The effect of HPβCD concentration was examined in intervals of (1.0 – 5.0) ×10-4 molL-1. It was observed that increasing in the concentration of HPβCD reagent caused a noticeable increase in the product absorbance when reaching 4.0×10-4 molL-1 as shown in Figure 8. Accordingly, 4×10-4 molL-1 of HPβCD reagent had been chosen as optimal concentration.

Figure 8: Effect of HPβCD reagent concentration on the reaction of LoP (6.0 mgL-1) with HPβCD, buffer solution (pH 1.0): 3.0 ml.
Effect of temperature
The impact of temperature was evaluated in the range of 25-40?C. The obtainable results as shown in Figure 9 indicated that the absorbance of LoP/HPβCD complex subsequently increase when increasing the temperature up to 35?C. Consequently, 35?C had been appointed as the optimum temperature.
The impact of temperature was evaluated in the range of 25-40?C. The obtainable results as shown in Figure 9 indicated that the absorbance of LoP/HPβCD complex subsequently increase when increasing the temperature up to 35?C. Consequently, 35?C had been appointed as the optimum temperature.

Figure 9: Effect of the temperature on the reaction of LoP (6.0 mgL-1) with HPβCD (4.0×10-4 molL-1), buffer solution (pH 1.0): 3.0 ml.
Effect of reaction time
The inclusion reaction was carried out in intervals of 5-25 min. It was recorded that the maximum absorbance of the reaction complex reached at 5 min and sharply decreased when increasing the time. Thus, 5 min had been designated as optimal time (data not shown.
The inclusion reaction was carried out in intervals of 5-25 min. It was recorded that the maximum absorbance of the reaction complex reached at 5 min and sharply decreased when increasing the time. Thus, 5 min had been designated as optimal time (data not shown.
Stoichiometry of the inclusion complex
Following the optimum conditions of the reaction between LoP and HPβCD, the stoichiometry was well performed by Job`s method and it was found to be 1:1. The result is shown in Figure 10.
Following the optimum conditions of the reaction between LoP and HPβCD, the stoichiometry was well performed by Job`s method and it was found to be 1:1. The result is shown in Figure 10.

Figure 10: Determination of the product formation by continuous variation method. VR: HPβCD (1.0×10-4 molL-1); VD: LoP (1.0×10-4 molL-1); VR+VD = 10 ml.
Method validation
The developed method validity wasthoroughlyexamined regarding to linearity, accuracy, precision, limits of detection (LOD) and quantification (LOQ), selectivity, and robustness according to ICH recommendations [45].
The developed method validity wasthoroughlyexamined regarding to linearity, accuracy, precision, limits of detection (LOD) and quantification (LOQ), selectivity, and robustness according to ICH recommendations [45].
Linearity and sensitivity
Under optimization conditions, the calibration graph was constructed by plotting the absorbance intensity of theinclusion reaction complex at 208 nm versus the concentration of LoP in mgL-1. The obtainable calibration curve was linear over the range 1.0-10 mgL-1. The regression equation had been reported from the data analysis with correlation coefficient of 0.9997 which indicating good linearity due to its high value and conformity of Beer`s law.On the other hand, method sensitivity is numerically represented in terms of molar absorptivity (ε, Lmol-1cm-1) and Sandell`s sensitivity which can be explicitly defined as the number of micrograms of elements which are in a column of solution having a cross section of 1 cm2 showing absorbance of 0.001 and expressed as µgml-1cm-2. The calculated values are abridged in Table 1.
Under optimization conditions, the calibration graph was constructed by plotting the absorbance intensity of theinclusion reaction complex at 208 nm versus the concentration of LoP in mgL-1. The obtainable calibration curve was linear over the range 1.0-10 mgL-1. The regression equation had been reported from the data analysis with correlation coefficient of 0.9997 which indicating good linearity due to its high value and conformity of Beer`s law.On the other hand, method sensitivity is numerically represented in terms of molar absorptivity (ε, Lmol-1cm-1) and Sandell`s sensitivity which can be explicitly defined as the number of micrograms of elements which are in a column of solution having a cross section of 1 cm2 showing absorbance of 0.001 and expressed as µgml-1cm-2. The calculated values are abridged in Table 1.
| Parameter | Value |
| LoP λmax, (nm) | 205 |
| LoP/HPβCD product λmax, (nm) | 208 |
| Beer`s law limits, (mgL-1) | 1.0 – 10 |
| Molar absorptivity, (Lmol-1 cm-1) | 4.8×104 |
| Sandell sensitivity, (µgcm-2) | 0.0050 |
| Limit of detection (LOD), (µgml-1) | 0.1327 |
| Limit of quantification (LOQ), (µgml-1) | 0.4022 |
| Regression Equation, Y* | |
| Intercept(a) | 0.0109 |
| Slope(b) | 0.0987 |
| Correlation coefficient(R2) | 0.9997 |
| Standard deviation | 0.0010 |
| Relative Standard deviation** (%) | 0.2507 |
*Y=a+bx, where Y is the absorbance, a the intercept, b the slope and x is the concentration in mgL-1.
** Average of three determinations
Table 1: Parameters for the performance of the proposed method.
** Average of three determinations
Table 1: Parameters for the performance of the proposed method.
Limits of Detection (LOD) and Quantification (LOQ)
The limit of detection (LOD) was determined by establishing the lowest concentration of LoP that can be readily detected and was measured according to ICH Q2(R1) recommendation, (LOD = 3.3 δ/S) where δ is the standard deviation of intercept of regression line of calibration curve and S is the slope. The limit of quantification (LOQ) was determined by evaluating the lowest concentration that can be calculated (LOQ = 10 δ/S). The recorded values of LOD and LOQ are summarized in Table 1.
The limit of detection (LOD) was determined by establishing the lowest concentration of LoP that can be readily detected and was measured according to ICH Q2(R1) recommendation, (LOD = 3.3 δ/S) where δ is the standard deviation of intercept of regression line of calibration curve and S is the slope. The limit of quantification (LOQ) was determined by evaluating the lowest concentration that can be calculated (LOQ = 10 δ/S). The recorded values of LOD and LOQ are summarized in Table 1.
Accuracy
The accuracy of the developed method was practically confirmed by performing recovery studies by addition of known amount of the standard pure LoP to pretreated formulations at three different concentrations within the linear range and each one in triplicate. The experimental results have been described in Table 2. As a result of these values, being within the acceptable range and having a smaller relative error, it was obvious that the method is extremely accurate for quantitative estimation of LoP in tablet dosage formulations.
The accuracy of the developed method was practically confirmed by performing recovery studies by addition of known amount of the standard pure LoP to pretreated formulations at three different concentrations within the linear range and each one in triplicate. The experimental results have been described in Table 2. As a result of these values, being within the acceptable range and having a smaller relative error, it was obvious that the method is extremely accurate for quantitative estimation of LoP in tablet dosage formulations.
| LoP Taken (mgL-1) | Added Sample content (mgL-1) | Found (mgL-1) | Recovery (% ± RSD)* | %RE |
| 2 | 2 | 4.18 | 104.50 ±0.14 | 0.04 |
| 4 | 2 | 6.32 | 105.30 ± 0.09 | 0.05 |
| 6 | 2 | 8.38 | 104.81 ± 0.07 | 0.05 |
*Average of three determinations
Table 2: Recovery studies for the determination of LoP by the proposed method.
Table 2: Recovery studies for the determination of LoP by the proposed method.
Precision
The precision of the proposed method was carefully evaluated by analyzing three different concentrations of LoP working standards. For intra-day precision, three successive replicate determinations of each concentration were determined within the same day following the general recommended procedure. The inter-day precision was carried out over three successive days with the same concentrations of LoP. The studied results are represented in Table 3, the calculated relative standard deviations which were less than 1.0 and the smaller relative error calculated indicated the good reproducibility of the developed method. So this precision is adequately required for the routine analysis of LoP in quality control laboratories.
The precision of the proposed method was carefully evaluated by analyzing three different concentrations of LoP working standards. For intra-day precision, three successive replicate determinations of each concentration were determined within the same day following the general recommended procedure. The inter-day precision was carried out over three successive days with the same concentrations of LoP. The studied results are represented in Table 3, the calculated relative standard deviations which were less than 1.0 and the smaller relative error calculated indicated the good reproducibility of the developed method. So this precision is adequately required for the routine analysis of LoP in quality control laboratories.
| LoP Taken (mgL-1) | Intra-day | Inter-day | ||||
| Found (mgL-1) | Recovery (% ±RSD)* | %RE | Found (mgL-1) | Recovery (%±RSD)* | %RE | |
| 4 | 4.22 | 105.43% ± 0.14 | 0.05 | 4.24 | 105.94 ± 0.14 | 0.06 |
| 6 | 6.29 | 104.90% ± 0.09 | 0.05 | 6.20 | 103.27 ± 0.25 | 0.03 |
| 8 | 8.27 | 103.37% ± 0.07 | 0.03 | 8.08 | 101.05 ± 0.13 | 0.01 |
*Average of three determinations
Table 3: Evaluation of intra-day and inter-day precision.
Table 3: Evaluation of intra-day and inter-day precision.
Selectivity
The selectivity of the proposed method had been examined by adding a minimal amount of known excipients such as starch, magnesium stearate, talc, and microcrystalline cellulose which are used in the dosage forms to investigate the interference. The analysis of these prepared samples following the recommended method revealed that the recovery values as shown in Table 4 had proved that there was no interference with these excipients, so the developed method is selective.
The selectivity of the proposed method had been examined by adding a minimal amount of known excipients such as starch, magnesium stearate, talc, and microcrystalline cellulose which are used in the dosage forms to investigate the interference. The analysis of these prepared samples following the recommended method revealed that the recovery values as shown in Table 4 had proved that there was no interference with these excipients, so the developed method is selective.
| No. | Excipient | Recovery (%± RSD)* |
| 1 | Starch | 105.92± 0.09 |
| 2 | Magnesium Stearate | 102.37± 0.26 |
| 3 | Talc | 105.52± 0.16 |
| 4 | Micro-crystalline Cellulose | 100.74± 0.36 |
*Average of three determinations
Table 4: Investigation of LoP in the presence of the excipients.
Table 4: Investigation of LoP in the presence of the excipients.
Robustness
The robustness had been investigated by evaluating the major influence of the slight variation on its analytical performance. In the experiments, one parameter was changed while others were kept unchanged and the recovery percentage was calculated each time. It was reported that the tiny variation in method variables did not significantly affect the procedure. So the recovery values that are shown in Table 5 approved the reliability of the developed method during the routine analysis of LoP.
The robustness had been investigated by evaluating the major influence of the slight variation on its analytical performance. In the experiments, one parameter was changed while others were kept unchanged and the recovery percentage was calculated each time. It was reported that the tiny variation in method variables did not significantly affect the procedure. So the recovery values that are shown in Table 5 approved the reliability of the developed method during the routine analysis of LoP.
| Parameter | Modification | Recovery (% ± RSD)* |
| pH | 0.95 | 102.71 ± 0.10 |
| 1.50 | 102.09 ± 0.10 | |
| Volume of buffer(ml) | 2.50 | 102.76 ± 0.10 |
| 3.50 | 103.44 ± 0.10 | |
| HPβCD Concentration (molL-1) |
0.0035 | 103.83 ± 0.17 |
| 0.0045 | 100.00 ± 0.34 | |
| Temperature (?C) | 30.00 | 102.93 ± 0.10 |
| 40.00 | 102.37 ± 0.26 | |
| Time(min) | 4.50 | 104.79 ± 0.09 |
| 5.50 | 104.56 ± 0.09 |
*Average of three determinations.
Table 5: Robustness of the proposed method.
Table 5: Robustness of the proposed method.
Method Application
The proposed method was successfully applied to the accurately determination of LoP in some pharmaceutical formulations. The practical results as shown in Table 6 strongly indicated the high accuracy of the developed method and can be easily used for the analysis of LoP in quality control laboratories, since the recovery obtainable values were satisfactory.
The proposed method was successfully applied to the accurately determination of LoP in some pharmaceutical formulations. The practical results as shown in Table 6 strongly indicated the high accuracy of the developed method and can be easily used for the analysis of LoP in quality control laboratories, since the recovery obtainable values were satisfactory.
| Tablet brand name | Label claim mg/tablet | Proposed method found (% ± RSD)* |
| Nilozaar | 50 | 104.62 ± 0.09 |
| Amilosan | 50 | 103.61 ± 0.10 |
*Average of three determinations.
Table 6: Results of assay of tablet formulations for the proposed method.
Table 6: Results of assay of tablet formulations for the proposed method.
Conclusion
The developed method was found to be simple, accurate, sensitive, economical, and easy. Such are the reproducible experimental results regarding the UV spectrophotometric method that it be directly applied to the routine quality control analysis of LoP in pharmaceutical dosage formulations.
References
- SS Hassan, SFA Zaidi, I Tariq, MT Ansari. (2013). Development and validation of analytical method for Losartan-copper complex using uv-vis spectroscopy. Trop J Pharm Res, 12(3): 407-411.
- PR Patil, SU Rakesh, PN Dhabale, KB Burade. (2009). Simultaneous uv spectrophotometric method for estimation of losartan potassium and amlodipine besylate in tablet dosage form. Asian J. Research Chem., 2(1): 183-187.
- ABWE Mohammed, EH Rudwan. (2015). Development and validation of an uv derivative spectrophotometric determination of losartan potassium in tablet. Der Pharma Chemica, 7(12): 175-180.
- L Latheeshjlal, P Parthiban, V Alagarsmy, M Sunil, JV Mahul, TR Mohan. (2010). Spectrophotometric determination of losartan potassium and its dosage form by bromothymolblue and phosphate buffer. E-J. Chem, 7(1): 320-324.
- N Rahman, MR Siddiqui, SNH Azmi. (2006). Development and validation of kinetic spectrophotometric method for the determination of losartan potassium in pure and commercial tablets. J. Chin. Chem. Soc., 53: 735-743.
- KV Lalitha, R Kiranjyothi, B Padma. (2013). UV spectrophotometric method development and validation for the determination of atenolol and losartan potassium by q-analysis. Int. Bull. Drug Res, 3(4): 54-62.
- KS Rao, K Srinivas. (2010). RP-HPLC method for the determination of losartan potassium and ramipril in combined dosage form. Indian J. Pharm. Sci., 72(1): 108-111.
- ADA Aninon, RSR Binos, KMM Brizuela, MC Corpuz, WJE De luna, RPS Del Rosario, et al. (2014). Spectrophotometric determination of losartan potassium in tablets. Sci. Diliman, 26(1): 41-52.
- M Masih, A Mittal, N BC. (2014). Spectrophotometric simultaneous estimation of amlodipine besylate and losartan potassium in tablet dosage forms. Asian J Pharm Clin Res, 7(4): 61-63.
- OC Lastra, IG Lemus, HJ Sanchez, RF Perez. (2003). Development and validation of an uv derivative spectrophotometric determination of losartan potassium in tablets. J Pharmaceut Biomed, 33(2): 175- 180.
- IA Darwish. (2005). Analytical study for the charge transfer complexes of losartan potassium. Anal. Chim. Acta, 549: 212-220.
- DD Tsvetkova, SA Ivanova. (2018). Application of uv-spectrophotometric method for the determination of losartan potassium in tablets. Indo Am. J. P. Sci, 5(8): 1-9.
- WX De Paula, AML Denadai, MM Santoro, ANG Braga, RAS Santos, RD Sinisterra. (2011). Supramolecular interactions between losartan potassium and hydroxypropyl-β-cd: esi mass-spectrometry, nmr techniques, phase solubility, isothermal titration calorimetry and anti-hypertensive studies. Int. J. Pharm, 404: 116-123.
- A Magnúsd?ttir, M Másson, M Loftsson. Cyclodextrins. (2002). J. Incl. Phenom. Macroc. Chem. 44: 213-218.
- H Xie, HY Wang, LY Ma, Y Xiao, J Han. (2005). Spectrophotometric study of the inclusion complex between β-cyclodextrin and dibenzoyl peroxide and its analytical application. Spectrochim Acta A, 62: 197-202.
- SB Carneiro, FIC Duarte, L Heimfath, JDeSS Quintans, LJ Quintans-Junior, VFDaV Junior, et al. (2019). Cyclodextrin-drug inclusion complexes: in vivo and vitro approaches. Int. J. Mol. Sci., 20(3): 642-664.
- WJ Shieh, AR Hedges. (1996). Properties and applications of cyclodextrins. J. Macromol. Sci. A, 33(5): 673-683.
- W Misiuk, E Jasiuk. (2014). Study of the inclusion interaction of hp-γ-cyclodextrin with bupropion and its analytical application. J. Mol. Struct, 1060: 272-279.
- T Loftsson, D Duchêne. (2007). Cyclodextrins and their pharmaceutical applications. Int. J. Pharm., 329: 1-11.
- Y Inoue, T Hakushi, Y Liu, LH Tong, BJ Shen, DS Jin. (1993). Thermodynamics of molecular recognition by cyclodextrins. 1. calorimetric titration of inclusion complexation of naphthalenesulfonates with α-, β-, and γ-cyclodextrins enthalpy-entropy compensation. J. Am. Chem. Soc., 115(2): 475-481.
- P Saokham, C Muankaew, P Jansook, T Loftsson. (2018). Solubility of cyclodextrins and drug/cyclodextrin complexes. Molecules, 23(5): 1161-1175.
- M Fermeglia, M Ferrone, A Lodi, S pricl. (2003). Host-guest inclusion complexes between anticancer drugs and β-cyclodextrin: computational studies. Carbohydr. Polym., 53: 15-44.
- ME Brewster, T Loftsson. (2007). Cyclodextrins as pharmaceutical solubilizers. Adv Drug Deliv Rev. 59: 645-666.
- NM Diez, AM De La Pena, MCM Garcia, DB Gil, FC Canada. (2007). Fluorimetric determination of sulphaguanidine and sulphamethoxazole by host-guest complexation in β-Cyclodextrin and partial least squares calibration. J. Fluoresc., 17: 309-318.
- AM Radi, SH Eissa. (2011). Voltametric and spectrophotometric studies on the inclusion complex of glipizide with β -CD. Eurasian J .Anal. Chem. 6(1): 13-21.
- T Aree, S Jongrungruangchok. (2018). β -Cyclodextrin encapsulation elevates antioxidant capacity of tea: a closing chapter on non-epicatechins, atomistic insight from X-ray analysis, DFT calculation and DPPH assay. Carbohydr. Polym., 194: 24-33.
- LJ Yang, B Yang, W Chen, R Huang, SJ Yan, J Lin. (2010). Host-guest system of nimbin and β -Cyclodextrin or its derivatives: preparation, characterization, inclusion mode, and solubilization. J. Agric. Food Chem., 58: 8545-8552.
- Y Zhang, YX Zhu, GL Huang, F Ren, FL Zheng, SJ Kim. (2001). Room temperature phosphorescence from inclusion complex of β -cyclodextrin and 1-bromonaphthalene in the presence of phenol and 1-butanol. Bull. Korean Chem. Soc. 22(12): 1397-1399.
- M Linar, MM Bertorello, M Longhi. (2000). Preparation and characterization of solid complexes of naphtoquinone and hydroxypropyl- β -cyclodextrin. Molecules, 5: 342-344.
- M Fermeglia, M Ferrone, A Lodia, S Pricl. (2003). Host-guest inclusion complexes between Anticancer Drugs and β-cyclodextrin: Computational studies, Carbohydr. Polym. 53: 15-44.
- JB Chao, HB Tong, SP Huang, DS Liu. (2005). Preparation and study on the solid inclusion complex of sparfloxacin with β-cyclodextrin. Spectrochim. Acta-Part A: Mole. Biomole. Spectra. 60: 161-166.
- NFA Dsugi, AA Elbashir. (2015). Supramolecular interaction of moxifloxacin and β-cyclodextrin spectroscopic characterization and analytical application. Spectrochim. Acta A: Mole. Biomole. Spectr., 137: 804-809.
- NFA Dsugi, AA Elbashir, HY Aboul-Enein. (2014). Supramolecular study on the interaction between ofloxacin and methyl Β-cyclodextrin by fluorescence spectroscopy and its analytical application. J Fluoresc, 24(2): 355-361.
- D Meng, X Hao, S Huang. (2004). Preparation and study on the novel solid inclusion complex of ciprofloxacin with HP- β –cyclodextrin. Spectrochim. Acta-Part A: Mole Biomole. Spectr., 60: 729-734.
- AA Elbashi, FO Suliman. (2011). Computational modeling of capillary electrophoretic behavior of primary amines using dual system18-crown-6 and β -cyclodextrin. J Chromatogr A, 1218: 5344-5351.
- AA Elbashir, B Saad, ASM Ali, MI Saleh, HY Aboul-Enein. (2010). Recent developments of enantioseparations for fluoroquinolone drugs using liquid chromatography and capillary. Curr. Pharmaceut. Anal., 6: 246-255.
- AA Elbashir, FEO Suliman, B Saad, HY Aboul-Enein. (2010). Capillary electrophoretic separation and computational modeling of inclusion complexes of β -CD and 18-crown-6 ether with primaquine and quinocide. Biomed. Chromatogr., 24: 393-398.
- AA Elbashir, NFA Dsugi, TOM Mohamed, HY Aboul-Enein. (2014). Spectrofluorometric analytical application of cyclodextrin. Luminescence. 29: 1-7.
- M Chen, G Diao, E Zhang. (2006). Study of inclusion complex of β-cyclodextrin and nitrobenzene. Chemosphere, 63: 522-529.
- S Shaomin, Y Yu, P Jinghao. (2002). Study on molecular recognition of para-aminobenzoic acid species by α-, β-, and hydroxypropyl-β-cyclodextrin. Analytical Chimica Acta, 458(2): 305-310.
- JMB Sendra, EA Lopez, AMG Campana, LC Rodriguez. (2003). Data analysis in the determination of stoichiometries and stability constants of complexes’, Anal. Sci., 19: 1431-1439.
- SJ Sonawane, KP Chaudhari, UT Jadhao, VM Thakare, BW Tekade, VR Patil. (2014). Design, development and evaluation of press coated tablets of an antihypertensive drug. Sch. Acad. J. Pharm., 3(2): 191-196.
- I Tadwee, S Shahi. (2018). Formulation development of losartan potassium immediate release tablets and process optimization using sedem expert system. J App Pharm Sci, 8(2): 33-43.
- AM Umakanthareddy, J Sreeeamulu, S Punna. (2012). Formulation development of losartan potassium microspheres using natural polysaccharides and their in-vitro evaluation. Res J Pharm Biol Chem Sci, 3(2): 725-734.
- Validation of analytical procedures, text and methodology (ICH Q2(R1), complementary guideline on methodology, (2005).
Citation: Hasna Ahmed Abdelhadi and Abdalla Ahmed Elbashir. (2020). Spectrophotometric Method Development and Validation for the Determination of Losartan Potassium in Pharmaceutical Formulations using Hydroxypropyl-β-cyclodextrin. Journal of Pharmacy and Drug Development 2(1).
Copyright: © 2020 Abdalla Ahmed Elbashir. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.